Théorie de Lewis
1 Concept accepteur-donneur
1.1 Les théories de l’acido – basicité
a) Théorie d’Arrhenius :
Selon Arrhenius un acide est une entité chimique qui libère en solution des protons H+.
Exemples : HCl, HNO3, H2SO4.
Une base au sens d’Arrhenius est une entité chimique qui libère en solution des ions hydroxydes OH-.
Exemples : NaOH, NH4OH, Ca(OH)2.
b) Théorie de Brönsted :
Un acide de Brönsted est un acide d’Arrhenius.
Une Base de Brönsted est une entité chimique qui capte en solution des protons H+.
Exemple :
![]()
Quand un acide au sens de Brönsted est fort il se dissocie totalement en solution.
![]()
Quand un acide au sens de Brönsted est faible il se dissocie partiellement en solution.
![]()
La théorie de Brönsted peut être reformulée ; en milieux aqueux (on dit aussi théorie de Brönsted – Lowry)
Un acide est une substance ; pas nécessairement neutre électriquement ; qui est capable de transférer ou céder son proton à une autre molécule.
![]()
Une base est une substance ; pas nécessairement neutre électriquement ; qui est capable d’accepter un proton d’une autre molécule.
![]()
Si on considère un acide faible AH ; il se dissocie pour donner H+ et A- base conjuguée de l’acide AH. On définit la constante d’acidité du couple (AH/A-) par l’expression :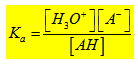
; pKa = - log Ka. Plus Ka est grand ; plus pKa est petit et plus l’acide est fort.
- Expression générale donnant le pH d’une solution :
Supposons une solution d’acide faible AH de concentration C de constante d’acidité Ka. On y ajoute n’ moles de son sel ANa.
![]()
La conservation de la quantité de matière donne : [AH] + [A-] = C + C’
L’électroneutralité de la solution donne : [H3O+] + [Na+] = [A-] + [OH-]
[H3O+] + C’ = [A-] + [OH-] d’où : [A-] = C’ + [H3O+] - [OH-]
[AH] = C - [H3O+] + [OH-]
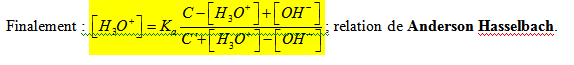
- Cas d’une solution d’acide faible :
Il n’y a pas de sel donc C’ = 0 ; les ions OH- ne proviennent que de la dissociation de l’eau ; en plus l’acide est faible donc [H3O+] est négligeable devant C ce qui donne : [H3O+]2 ≈ KaC
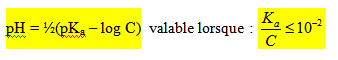
- Cas d’une solution de base faible :
Dans ce cas C = 0 et les ions H30+ proviennent uniquement de la dissociation de l’eau et sont négligeables ; de plus c’est une base faible et [OH-] est faible devant C’ ce qui donne :
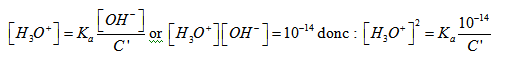
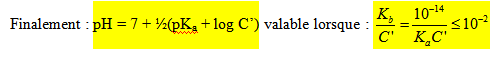
- Cas d’une solution tampon :
Une solution tampon est une solution dont le pH varie peu ou presque pas quand on ajoute de l’acide ou de la base ou encore de l’eau en quantité modérée. C’est le cas d’une solution contenant un acide et sa base conjuguée. Dans l’expression de Anderson Hasselbach on néglige [H3O+] et [OH-] devant C et C’ d’où
![]()
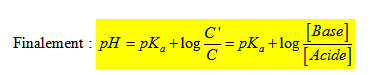
- Cas du mélange de deux systèmes acide – base :
Considérons un sel d’acide faible en solution ; l’acétate d’ammonium par exemple ; on a l’équilibre suivant :
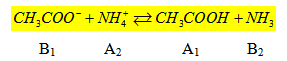
Nous avons un mélange de deux systèmes acide – base. Nous pouvons écrire en appelant K1 et K2 les constantes d’acidité des couples (A1/B1) et (A2/B2).
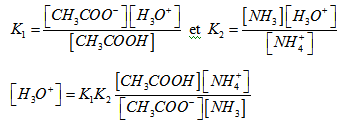
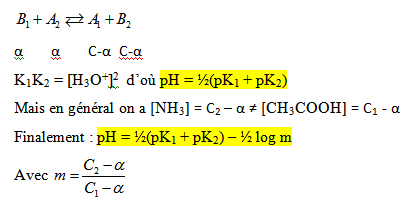
- Cas des ampholytes :
Une solution d’ampholytes est équivalente à un mélange de deux systèmes acide – base. C’est l’exemple du cas des sels obtenus par neutralisation de la première acidité d’un polyacide faible. Exemple de l’hydrogénosulfite de sodium NaHS. Na+ est indifférent à l’eau.
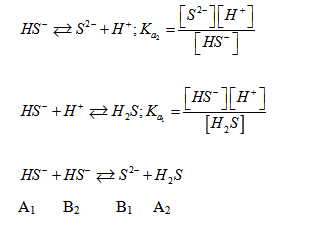

- Dissociation des sels :
Tous les sels se dissocient totalement sauf un sel d’acide faible et de base faible qui donnent un mélange de deux systèmes acide – base.
- Dosage d’un acide fort par une base forte :
L’équivalence acido – basique est obtenue lorsque le nombre de moles de base versée CbVb est égal au nombre de moles d’acide initialement contenues dans le bécher CaVa.
Plusieurs étapes correspondant à des volumes de base donnés sont à distinguer :

- Dosage d’un acide faible par une base forte :
Vb = 0
On a un mélange tampon ; au point équivalent on a un sel d’acide faible et de base forte qui se dissocie totalement et on a une solution de base faible.
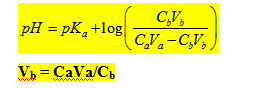
Au point équivalent tout l’acide AH a été neutralisé en base faible A-. Le pH est donc celui d’une solution de base faible ;
c) Théorie de Lewis :
L’acide est un accepteur d’électrons et la base un donneur d’électrons.
L’acide de Lewis est une entité chimique ayant une vacance de doublet c’est – à – dire qu’il peut recevoir un doublet électronique.
Exemple : BF3. Le bore a six électrons sur sa couche externe et compte tenue de la règle de l’octet ; il peut avoir un autre doublet pour s’entourer de huit électrons. BF3 a donc une vacance électronique d’où il est acide par rapport au bore.
Une base de Lewis est une entité chimique ayant un ou plusieurs doublets libres sur un de ses atomes.
Exemple NH3 est une base de Lewis par rapport à l’azote.
Un acide de Lewis reçoit grâce à sa vacance électronique un doublet venant d’une base de Lewis par liaison dative. Un acide de Lewis réagit avec une base de Lewis pour donner un complexe de coordination.
Pour écrire la formule des complexes, on écrit d’abord le métal puis les ligands qu’on met entre parenthèses s’ils comportent plus d’un atome, le tout entre crochets et en dehors de ceux – ci la charge du complexe exemple [Ni(CN)4]2-.
Les métaux non transitionnels et les métaux transitionnels sont des acides de Lewis dans la plupart de leurs états d’oxydation. Ils peuvent recevoir un doublet de chacun des ligands qui les entourent pour donner un complexe de coordination.
Exemple : Zn2+ + 4 Cl- → [ZnCl4]2-
- Les effets stériques :
En faisant réagir un acide et une base on doit toujours avoir à l’esprit la distorsion que subit chaque molécule dans sa configuration au sein du composé formé. La présence de gros substituants peut affecter la stabilité du composé formé. Cette stabilité est diminuée par le fait que l’acide et la base ne peuvent se rapprocher assez près l’un de l’autre pour que le recouvrement des orbitales soit maximal.
Considérons le complexe formé entre le triéthylamine et le triméthylborane :
Et3N :→ BMe3
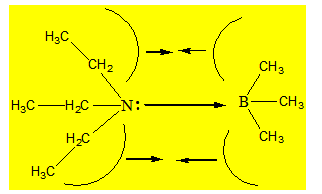

Ce phénomène de gêne frontale est appelé F – strain et il peut avoir une influence considérable sur la stabilité des composés formés puisque les cônes de révolution des gros substituants peuvent avoir une base très large. Si on compare ce complexe avec NH3 – BF3 ; la distance entre l’azote et le bore est plus petite que celle du complexe précédent. L’approche de l’azote dans de ce dernier complexe est gênée par la rotation libre des gros substituants autour des liaisons σ qui décrivent des cônes de révolution. Il arrive un moment où ces cônes interfèrent et il ne sera plus possible aux atomes accepteur et donneur de se rapprocher davantage l’un de l’autre.
- Back – strain ou B – strain :
Si on considère la triméthylamine N(CH3)3 et la trisilylamine N(SiH3)3 ces deux molécules d’après V.S.E.P.R. devraient avoir des géométries pyramidales ; on voit que la première est pyramidale alors que la deuxième est plane. On explique cette anomalie en considérant que dans N(SiH3)3 on devrait avoir une pyramide mais compte tenu de la grosseur des substituants silyles il y a de fortes répulsions entre eux et la molécule n’est pas stable dans cette configuration. Pour se stabiliser la molécule entame un processus qui lui permet d’éloigner les substituants les uns des autres et pour cela elle s’écrase sur elle – même suivant la normale au plan des trois silicium passant par l’atome d’azote. Quand ce processus d’écrasement sur elle – même est terminé, la molécule est plane mais on ne peut pas expliquer la géométrie plane de cette molécule sans qu’il y ait un back – bonding N – Si (recouvrement de type latéral pour donner une liaison π). On considère comme état excité de l’azote :
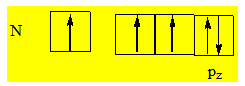
L’orbitale pz de l’azote est parallèle à l’orbitale dyz du silicium et on aura un recouvrement de type latéral ; ce qui donne un back – bonding pπdπ. Il ya un phénomène de résonance pour la symétrie de la molécule. Dans cet exemple on part d’une hybridation sp3 et on arrive à une hybridation sp2 et les angles sont standards. Le processus de B – strain n’est pas nécessairement un processus faisant passer une molécule d’un angle standard à un autre angle standard. La molécule cherche à éloigner les substituants les uns des autres pour avoir une bonne stabilité. Si en arrivant à un angle de 115° elle minimisait ces répulsions ; elle n’aurait pas besoin de prolonger le processus de B – strain vers un angle de 120° et elle se serait tout simplement arrêtée à 115°. Tous les états intermédiaires entre 109° et 120° correspondants à une valeur α sont un état d’équilibre possible. Ce processus de B – strain s’accompagne toujours d’un back – bonding. Le back – bonding est complet quand le B – strain passe d’un angle standard à un autre angle standard. La présence d’une ou de plusieurs paires libres sur l’atome central et d’une ou de plusieurs orbitales vides sur l’acide de Lewis sont des conditions nécessaires mais non suffisantes pour avoir un B – strain. Il peut arriver que certaines molécules possèdent un doublet libre sur l’élément central et des orbitales vides sur l’acide de Lewis et ne pas subir de B – strain.
Le rôle de l’élément central est très important dans le B – strain. Si nous considérons la molécule de trisilylphosphine P(SiH3)3 ; elle est beaucoup plus basique que la trisilylamine N(SiH3)3 ce qui veut dire que le doublet du phosphore est totalement libéré d’interactions de type back – bonding autrement dit malgré la présence d’une paire libre et d’orbitales d vides ; il n’y a pas de B – strain. En fait le phosphore est plus gros que l’azote donc les SiH3 sont plus éloignés les uns des autres dans la trisilylphosphine que dans la trisilylamine. Les répulsions étant faibles dans la trisilylphosphine la molécule n’a pas besoin de s’imposer un B – strain et elle reste pyramidale et conserve sa paire libre alors que la trisilylamine engage un processus de B – strain pour se stabiliser et ce phénomène s’accompagne de back – bonding utilisant le doublet libre de l’azote. D’où sa basicité diminue.
d) Nomenclature des composés binaires :
L’élément électropositif est placé en tête. Le nom du constituant n’est pas modifié sauf dans le cas des métaux possédant deux valences. On met les suffixes « eux » et « ique » pour la valence la moins élevée et la plus élevée respectivement. Le nom du constituant électronégatif est suivi de la terminaison « ure ». On l’énonce en tête. Exemple : FeCl2 chlorure ferreux ; FeCl3 chlorure ferrique.
Dans le cas où on a plus de 2 valences on utilise les préfixes « hypo » et « per ».
Cl2O anhydride hypochloreux
Cl2O3 anhydride chloreux
Cl2O5 anhydride chlorique
Cl2O7 anhydride perchlorique
Les acides donnant des sels en « ure » ont leur terminaison en « hydrique ».
Exemple : acide chlorhydrique HCl donne l’ion chlorure.
Les acides donnant des sels en « ite » ont leur terminaison en « eux »
Exemple : Acide sulfureux H2SO3 donne l’ion sulfite
Acide nitreux HNO2 donne l’ion nitrite.
Les acides à terminaison « ique » donnent des sels en « ate ».
Exemple : acide sulfurique donne l’ion sulfate.