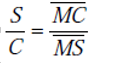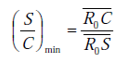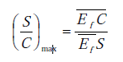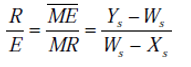cours sur le chapitre 2
Le contenu du cours est disponible ci-dessous en ligne.
| Site: | Touch By SukaJanda01 |
| Cours: | CHIM 4212 :Méthode d'analyses chimiques |
| Livre: | cours sur le chapitre 2 |
| Imprimé par: | Visiteur anonyme |
| Date: | mardi 17 juin 2025, 02:10 |
Table des matières
- 1 Cours de Microanalyse
- 1.1 GÉNÉRALITÉS SUR LES OPÉRATIONS DE SÉPARATION
- 1.2 GÉNÉRALITÉS SUR LES OPÉRATIONS DE SÉPARATION
- 1.3 Courbe d’équilibre
- 1.4 Force motrice du procédé de transfert de masse
- 1.5 Analyse d'échantillon
- 1.6 La Chromatographie sur Couche Mince
- 1.7 Principe de la technique.
- 1.8 CCM
- 1.9 CCM Calcul du Rf
- 1.10 Principe de la CCM
- 1.11 Procéder à une identification en CCM
- 1.12 Procédés d’extraction liquide-liquide
- 1.13 Principe de l’extraction liquide-liquide :
- 1.14 Différents types d’extraction
- 1.15 Extraction à contre-courant
- 1.16 Sélectivité du solvant
- 1.17 Données d’équilibre liquide-liquide
- 1.18 Règle des mélanges: relation barycentrique
- 1.19 Expression des propriétés physico-chimiques
- 1.20 Système ternaire: Autres diagrammes
- 1.21 Diagramme de sélectivité
- 1.22 Calcul du nombre d’étages théoriques d’une colonne d’extraction liquide-liquide
1 Cours de Microanalyse
Chapitre sur la Microanalyse est destiné à la 1ère Année Master I (LMD)
Introduction aux méthodes d’analyse
Les méthodes d’analyses trouvent leur application dans des domaines extrêmement variés. Qu’il s’agisse de surveiller la qualité des produits alimentaires, de tester des moteurs dans l’industrie automobile, de commander des processus dans l’industrie chimique ou pharmaceutique, dans la médecine, la métallurgie ou la surveillance de l’environnement, partout des méthodes d’analyse sont mises en oeuvre pour commander des processus, pour garantir la qualité ou même pour prouver que l’on respecte les réglementations légales.
Il est indispensable pour appréhender un matériau de le caractériser, c'est-à-dire d'en analyser les propriétés. Il existe de nombreuses techniques de caractérisation des matériaux qui reposent sur différents principes physiques de base : les interactions rayonnement-matière, la thermodynamique et la mécanique.
Pour quoi on analyse ?
ALALYSE quantitative: dosage, rendement, % relatif.
Analyse quantitative : caractérisation, détection, identification.
On distingue deux méthodes d’analyse
Méthodes physiques : basées: sur les propriétés physiques ( Teb, viscosité, indice de réfraction
Méthodes Spectroscopiques: basées sur l’interaction matière /lumière.
Les principales techniques utilisant l'interaction rayonnement-matière sont :
microscopie :
o microscopie électronique à balayage (MEB) : elle est une technique de microscopie électronique basée sur le principe des interactions électrons-matière,
o microscopie électronique en transmission (MET) : on bombarde d'un faisceau d'électrons un échantillon mince principalement pour en obtenir la figure de diffraction,
o microscope optique : la simple et traditionnelle observation à l'échelle microscopique ;
rayons X : diffraction des rayons X, fluorescence X, radiographie ;
rayon de neutrons : diffraction de neutrons, neutronographie et activation neutronique.
En ce qui concerne les techniques faisant appel à la thermodynamique, l’analyse thermique, on retrouve la calorimétrie différentielle à balayage (DSC) et l'analyse thermogravimétrique (ATG). Voir aussi Analyse thermomécanique (TMA).
1.1 GÉNÉRALITÉS SUR LES OPÉRATIONS DE SÉPARATION
I.1. Généralités sur les opérations de séparation
I.1.1. Introduction
Les opérations de séparation des mélanges en leurs composants purs sont d’une grande importance industrielle. Elles ont rendu un grand service à l’humanité surtout dans l’industrie pétrochimique (fractionnement du pétrole).
Ce sont des processus de transfert de masse qui réalisent leurs objectifs par la création de deux ou plusieurs zones coexistantes qui diffèrent en température, en pression, en composition ou en état de phase.
I.1.2. Méthode de traitement d’un problème de séparation
Quelle est la méthode ou bien l’opération unitaire à mettre en œuvre pour réaliser une séparation ?
La distillation est une grande consommatrice d’énergie et les industriels ont alors envisagé d’autres méthodes de séparation (extraction-cristallisation- absorption……). Le choix de notre opération unitaire entraine un accroissement du coût qui est compensé à plus ou moins long terme par les économies d’énergie réalisées.
I.1.3. Classification des opérations de séparation
Ces opérations sont très répandues dans l’industrie, ou elles jouent un rôle très important dans le transfert de matière. On distingue :
a- La distillation
La distillation est un procédé de transfert de matière qui sert à séparer un mélange liquide en se basant sur la différence des températures d’ébullition des composants du mélange.
b- La condensation partielle et vaporisation flash
Quand le mélange à séparer contient des espèces qui diffèrent largement dans leur tendance à la vaporisation et à la condensation, une vaporisation ou bien une condensation partielle est l’opération adéquate pour accomplir la séparation désirée.
Ø Dans la condensation partielle, l’alimentation vapeur est partiellement condensée par déplacement de la chaleur.
Ø Dans la vaporisation flash, l’alimentation liquide est partiellement vaporisée par réduction de la pression.
1.2 GÉNÉRALITÉS SUR LES OPÉRATIONS DE SÉPARATION
c- L’absorption
L’absorption est un procédé physique de séparation des mélanges gazeux à l’aide de liquides absorbants. Pour que l’absorption soit réalisable, il faut que l’absorbant choisi ait une grande sélectivité, c’est-à-dire qu’il absorbe seulement l’élément qu’on veut déplacer de la phase gazeuse.
- Le constituant que l’on veut extraire est appelé : soluté
- Le gaz dans lequel il est porté est appelé: gaz porteur
- Le liquide dans lequel il se dissout est appelé : solvant
Il y a deux cas d’absorption :
Ø Absorption chimique: où le soluté entre en réaction avec le solvant, donc on obtient dans ce cas un composé chimique.
Ø Absorption physique: où la nature du corps ne change pas, ce qui permet la régénération du solvant et ainsi sa réutilisation.
d- L’extraction liquide-liquide
Basée sur le fait de la séparation d’un mélange liquide constitue de plusieurs composants en le traitant avec un autre liquide qu’on appelle dissolvant, une séparation basée sur la différence de solubilité dans le solvant des divers constituants. Les deux liquides constituent généralement une émulsion c’est-à-dire liquides non miscibles.
e- L’évaporation
C’est un procède physique de séparation des solutions par vaporisation du solvant. Les appareils permettant d’effectuer ce procède sont appelés évaporateurs.
f- Adsorption (fluide-solide)
Cette operation se manifeste lorsqu’on met en contact un fluide et un solide, elle n’est utilisée industriellement que dans les domaines assez restreints (séchage- traitement des huiles traitement
des gaz).
g- Cristallisation
Obtenue par abaissement la température du mélange, elle est très utilisée dans le domaine de la chimie minérale et dans la préparation des produits organiques.
h- Séchage
Dans ce cas, la matière se déplace de la phase solide à la phase vapeur.
1.3 Courbe d’équilibre
I.2. Courbe d’équilibre
C’est la courbe qui représente la relation des compositions des deux phases à l’état d’équilibre. On la détermine expérimentalement.
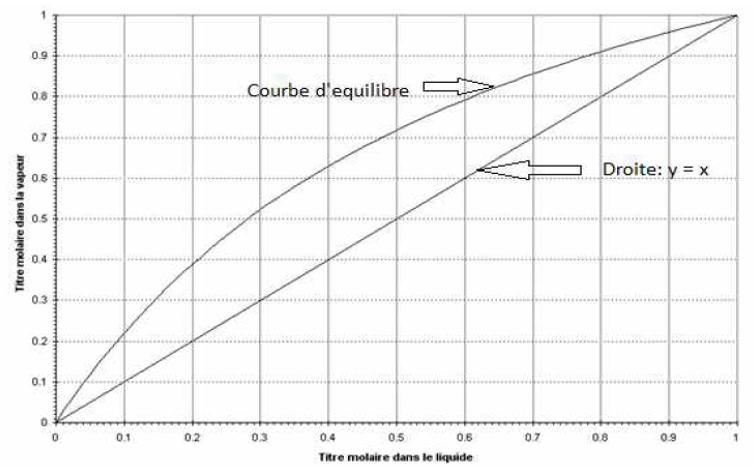
Figure I.1: Courbe d’équilibre du mélange binaire Toluène-Benzène
I.3. Courbe opératoire
C’est la courbe représentant la relation des compositions pratiques des deux phases. Cette courbe est considérée comme une droite (linéaire) : y = ax+b
Selon le cas, la courbe opératoire peut se trouver au-dessus ou en dessous de la courbe d’équilibre.
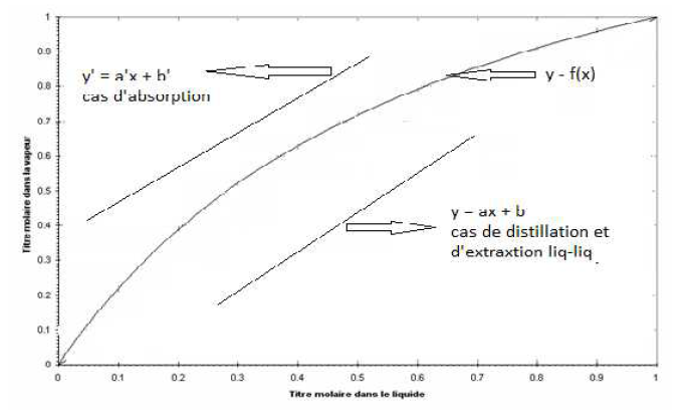
Figure I.2: Courbe opératoire
1.4 Force motrice du procédé de transfert de masse
I.4. Force motrice du procédé de transfert de masse
La force motrice du procédé de transfert de masse est la différence entre la composition d’équilibre et la composition pratique.
La force motrice n’est pas constante le long de l’appareil, c’est pourquoi dans les calculs, on utilise la force motrice moyenne : Δym et Δxm, y représentant la concentration du soluté en phase vapeur et x la concentration du soluté en phase liquide.
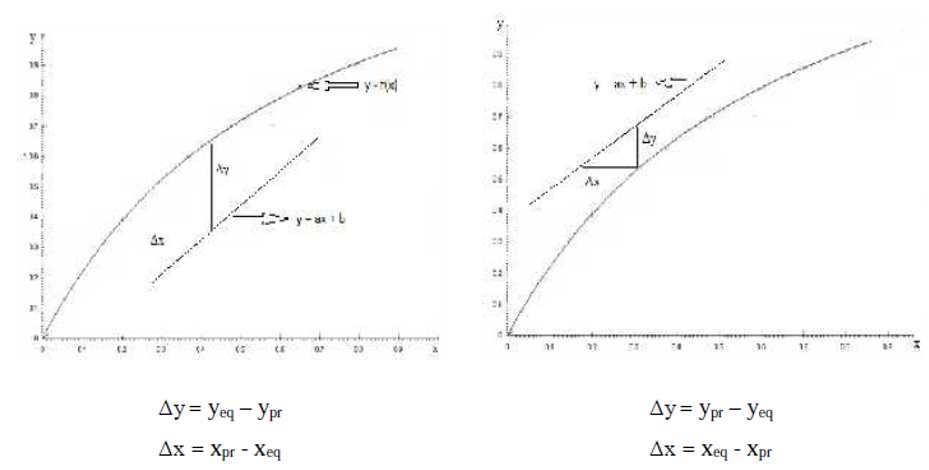
Figure I.3: Force motrice du procédé de transfert de masse
I.5. Transferts de matières
Les transferts de matières ont lieu d’une phase à une autre. Chaque type de transfert est désigne
de façon particulière comme cela est exprimé selon le tableau suivant :
Tableau I.1: Types de transferts
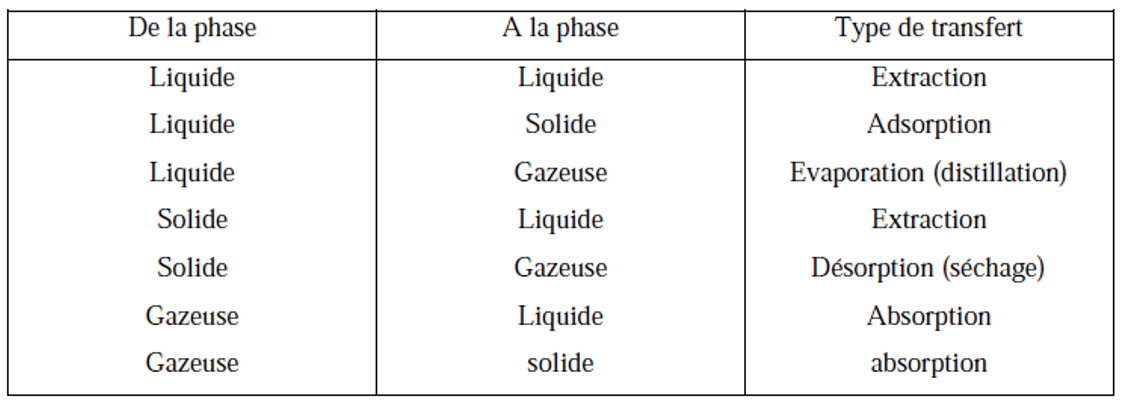
Le principe des transferts de matière est basé sur le fait que le composant qui est l’objet du transfert a une solubilité différente dans chacune des phases ; cela provoque le passage de ce composant de la phase où il est le moins soluble à celle où il l’est de façon appréciable.
1.6 La Chromatographie sur Couche Mince
La Chromatographie sur Couche Mince
- 1. Définition et appareillage.
La chromatographie sur couche mince (CCM) repose principalement sur des phénomènes d’adsorption : la phase mobile est un solvant ou un mélange de solvants, qui progresse le long d’une phase stationnaire fixée sur une plaque de verre ou sur une feuille semi-rigide de matière plastique ou d’aluminium.
Après que l’échantillon ait été déposé sur la phase stationnaire, les substances migrent à une vitesse qui dépend de leur nature et de celle du solvant.
Les principaux éléments d’une séparation chromatographique sur couche mince sont :
– la cuve chromatographique : un récipient habituellement en verre, de forme variable, fermé par un couvercle étanche.
– la phase stationnaire : une couche d’environ 0,25 μm de gel de silice ou d’un autre adsorbant est fixée sur une plaque de verre à l’aide d’un liant comme le sulfate de calcium hydraté (plâtre de Paris) l’amidon ou un polymère organique.
– l’échantillon : environ un microlitre (μl) de solution diluée (2 à 5 %) du mélange à analyser, déposer en un point repère situé au-dessus de la surface de l’éluant.
– l’éluant : un solvant pur ou un mélange : il migre lentement le long de la plaque en entraînant les composants de l’échantillon.
1.7 Principe de la technique.
2. Principe de la technique.
Lorsque la plaque sur laquelle on a déposé l’échantillon est placée dans la cuve, l’éluant monte à travers la phase stationnaire, essentiellement par capillarité. En outre, chaque composant de l’échantillon se déplace à sa propre vitesse derrière le front du solvant.
Cette vitesse dépend d’une part, des forces électrostatiques retenant le composant sur la plaque stationnaire et, d’autre part, de sa solubilité dans la phase mobile. Les composés se déplacent donc alternativement de la phase stationnaire à la phase mobile, l’action de rétention de la phase stationnaire étant principalement contrôlée par des phénomènes d’adsorption. Généralement, en chromatographie sur couche mince, les substances de faible polarité migrent plus rapidement que les composants polaires.
3. Applications de la CCM.
Lorsque les conditions opératoires sont connues, elle permet un contrôle aisé et rapide de la pureté d’un composé organique. Si l’analyse, réalisée avec divers solvants et différents adsorbants, révèle la présence d’une seule substance, on peut alors considérer que cet échantillon est probablement pur.
De plus, étant donné que la chromatographie sur couche mince indique le nombre de CHROMATOGRAPHIE SUR COUCHE MINCE 1 sur 4 composants d’un mélange, on peut l’employer pour suivre la progression d’une réaction.
4. Adsorbants et plaques chromatographiques.
Par ordre d’importance décroissante, les adsorbants employés en CCM sont : le gel de silice, l’alumine, le kieselguhr et la cellulose.
Les plaques vous seront fournies prêtes à l’emploi.
5. Choix de l’éluant.
L’éluant est formé d’un solvant unique ou d’un mélange de solvants. Un éluant qui entraîne tous les composants de l’échantillon est trop polaire ; celui qui empêche leur migration ne l’est pas suffisamment.
Choix de l’éluant dans le cas d’analyses :
– d’hydrocarbures : hexane, éther de pétrole ou benzène.
– de groupements fonctionnels courants : hexane ou éther de pétrole mélangés en proportions variables avec du benzène ou de l’éther diéthylique forment un éluant de polarité moyenne.
– de composés polaires : éthanoate d’éthyle, propanone ou méthanol.
1.8 CCM
6. Dépôt de l’échantillon.
L’échantillon est mis en solution (2 à 5 %) dans un solvant volatil, qui n’est pas forcément le même que l’éluant : on emploie fréquemment le trichlorométhane (chloroforme), la propanone ou le dichlorométhane. La solution est déposée en un point de la plaque situé à environ 1 cm de la partie inférieure.
Il est important que le diamètre de la tache produite au moment du dépôt soit faible ; idéalement, il ne devrait pas dépasser 3 mm. Ce sont généralement les dépôts les moins étalés qui permettent les meilleures séparations. Pour augmenter la quantité déposée, il est toujours préférable d’effectuer plusieurs dépôts au même point, en séchant rapidement entre chaque application plutôt que de déposer en une seule fois un grand volume d’échantillon qui produirait une tache plus large.
L’échantillon est déposé à l’aide d’une micropipette ou d’un tube capillaire en appuyant légèrement et brièvement l’extrémité de la pipette sur la couche d’adsorbant en prenant soin de ne pas le détériorer.
On vérifie l’identité des composants présumés d’un échantillon, en procédant à un dépôt séparé d’une solution de chacun d’eux puis à celui de leur mélange. Ces solutions témoins permettent de comparer la migration de chaque composé avec celle de l’échantillon à analyser.
7. Développement de la plaque.
Le développement consiste à faire migrer le solvant sur la plaque. Dans les analyses usuelles de laboratoire, le principal type de développement est la chromatographie ascendante : la plaque est placée en position verticale dans une cuve et le solvant qui en recouvre le fond monte par capillarité.
Le niveau de liquide est ajusté à environ 0,5 cm du fond de la cuve puis on introduit la plaque. Pendant le développement du chromatogramme, la cuve doit demeurer fermée et ne pas être déplacée.
Lorsque la position du front du solvant arrive à environ 1 cm de l’extrémité supérieure, la plaque est retirée de la cuve, le niveau atteint par le solvant est marqué par un trait fin, puis la plaque est séchée à l’air libre ou à l’aide d’un séchoir.
8. Révélation.
Lorsque les composants de l’échantillon analysé sont colorés, leur séparation est facilement observable sur la plaque ; dans le cas contraire, on doit rendre les taches visibles par un procédé de révélation. Les taches sont ensuite cerclées au crayon. Les méthodes usuelles de révélation sont les suivantes : radiations UV, fluorescence, iode , atomisation.
Sauf indications contraires, nous utiliseront les radiations UV. En exposant la plaque à une source de radiation UV, certains composés apparaissent sous forme de taches brillantes.
Si un indicateur fluorescent est incorporé à l’adsorbant, la plaque entière devient fluorescente lorsqu’elle est soumise à une radiation UV ; les composés y sont révélés sous forme de taches sombres.
1.9 CCM Calcul du Rf
9. Calcul de Rf (retarding factor ou rapport frontal).
di : distance parcourue par le composé (mesuré au centre de la tache)
ds : distance parcourue par le front du solvant
10. Description d’une analyse par CCM selon l’ordre chronologique.
CHROMATOGRAPHIE SUR COUCHE MINCE
Préparation de la cuve chromatographique.
– Introduire l’éluant ou le mélange de solvants.
– Ajuster le niveau à environ 0,5 cm du fond de la cuve.
– Garnir l’intérieur de la cuve d’un papier filtre imprégné d’éluant et plaqué contre les parois ; une ouverture est ménagée dans le filtre pour observer le développement du chromatogramme.
– Fermer le récipient (la cuve doit être saturée de vapeur de solvant)
Dépôt de l’échantillon sur la plaque.
– Procéder au nettoyage de la plaque si nécessaire.
– Dissoudre l’échantillon dans un solvant approprié en solution de 2 à 5 % .
– Déposer environ 0,5 ml de la solution en un point situé à 1 cm de l’extrémité inférieure de la plaque; le diamètre de la tache doit être d’environ 2 mm pour la disposition de plusieurs produits.
– Sécher à l’aide d’un séchoir ; éventuellement faire de nouvelles applications.
Développement du chromatogramme.
– Placer la plaque dans la cuve en position verticale.
– Refermer le récipient qui ne doit plus être déplacé.
– Lorsque le front du solvant se trouve à environ 1 cm de l’extrémité supérieure de la plaque, la retirer et marquer cette position. (le trait peut être tracé à l’avance et servir de repère pour arrêter l’élution).
Révélation et calcul de Rf.
– Sécher la plaque à l’aide d’un séchoir
– Révéler les taches sous une lampe U V
– Cercler les taches et pointer leur centre.
– Calculer les Rf
1.10 Principe de la CCM
Chromatographie sur couche mince (CCM)
Principe de la CCM
La chromatographie sur couche mince s’effectue généralement sur une fine couche de silice (phase stationnaire) déposée sur un support. Le mélange à étudié est ensuite posé à l’aide d’un capillaire à environ 1 cm du bord puis placé dans une cuve contenant l’éluant. Le niveau de l’éluant devant être en dessous du produit déposé. La cuve de chromatographie est ensuite refermée par un couvercle. L’éluant migre sur la plaque de silice par capillarité et entraîne les composés du mélange étudié. Si les vitesses de migration des composés sont différentes, ils seront séparés. La plaque de chromatographie est ensuite lue directement si les composés sont visibles, ou placé sous une lumière UV. Ils peuvent également être révélés en pulvérisant une solution d’acide sulfurique puis chauffé dans une étuve.
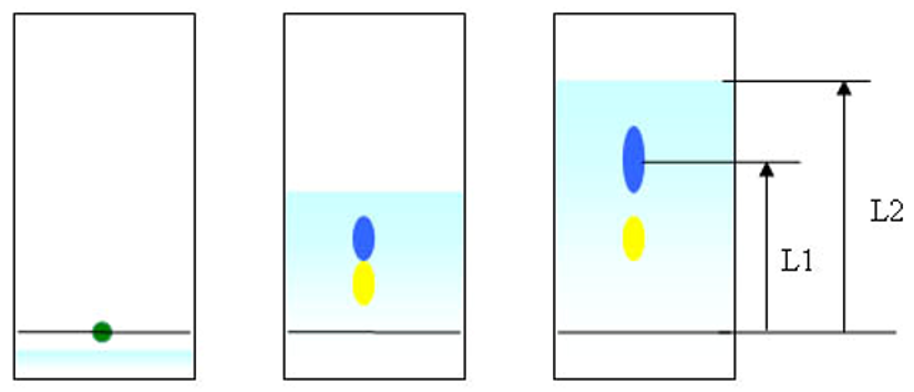
On détermine le ratio frontal Rf = L1/L2 étant le rapport entre la distance parcourue par le soluté divisé par la distance parcourue par le front du solvant.
Le principe de séparation des composés par CCM est proche de celle en HPLC. Le mélange est placé sur la plaque de silice à l'aide d'une pipette pasteur.
Le principal intérêt de la CCM est l'identification rapide des composés d'un mélange. En contre partie, l'analyse est uniquement qualitative et ne permet pas le dosage d'un composé.
1.11 Procéder à une identification en CCM
Pour identifier les composés d’un mélange, on effectue une CCM de ce mélange avec ceux des produits purs supposé présents, le tout sur la même plaque.
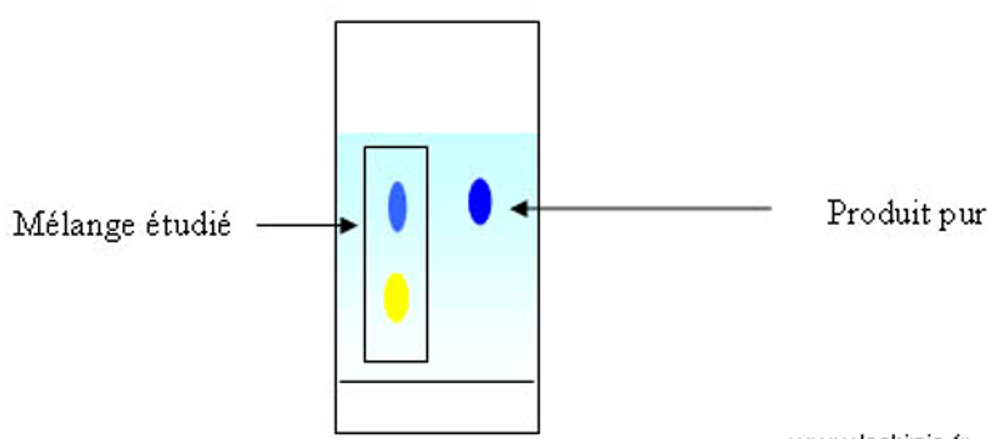
Le produit pur en « bleu » a le même Rf que celui du mélange à étudier. Cela confirme la présence du produit « bleu » dans le mélange étudié. Une confirmation peut être amenée à l’aide d’une 2ème élution dans un autre solvant en faisant pivoter la plaque de CCM. On utilise dans ce cas des plaques de forme carrée. On appelle cette méthode la CCM bidimensionnelle.
Exemple de CCM bidimensionnelle
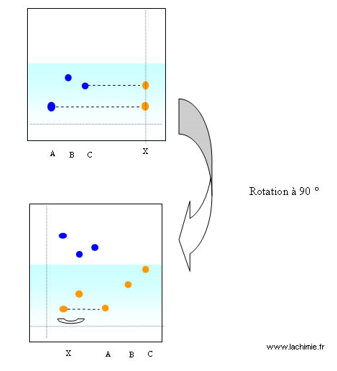
La double élution permet de montrer la présence du composé « A » dans le mélange. Le composé qui avait le même Rf dans la première élution que « C » est en réalité un autre produit.
1.12 Procédés d’extraction liquide-liquide
III.1. Terminologie
- Soluté : constituant à extraire
- Diluant: liquide contenant les solutés
- Solution: ensemble soluté + diluant
- Solvant: liquide destiné à extraire les solutés
- Extrait : phase issue de l’opération contenant les solutés extraits. Cette phase est riche en solvant.
- Raffinat : phase résiduelle épuisée en soluté. Cette phase est riche en diluant.
- Phase lourde : phase ayant la plus grande masse volumique.
- Phase légère : phase ayant la plus faible masse volumique.
- Phase aqueuse/Phase organique : ces termes sont liés à la nature du solvant et du diluant.
- Un azéotrope est un mélange liquide qui bout à température fixe en gardant une composition fixe.
III.2. Définition
L'extraction liquide/liquide (ou LLE) est l'une des techniques de séparation d'échantillons les plus anciennes. Elle permet, par un procédé physique, de purifier ou d'extraire des classes de composés (ou solutés) de la matrice (ou éluant) dans lequel ils sont solubilisés. Le solvant d'extraction ne doit pas être miscible avec l'éluant tout en solubilisant le maximum de solutés.
Un mélange binaire dont on veut effectuer la séparation est mis en contact avec un troisième liquide non miscible appelé solvant et, retenu pour sa capacité à extraire préférentiellement l'un des éléments du mélange. Après l'opération, on récupère deux phases séparées par décantation: l'extrait formé du solvant enrichi en soluté, et le raffinat, soit le mélange appauvri en soluté.
On distingue deux types d'extraction liquide-liquide:
- L'extraction par échange d'ions: la phase d'alimentation s'enrichit d'une espèce contenue dans le solvant pendant que le soluté passe dans le solvant. Elle repose sur une réaction chimique et l'échange de cations et anions.
- L'extraction non-compensée: simple transfert de molécules ou d'agrégats d'ions globalement neutres. L'extraction peut être faite avec un réactif solvatant, permettant un transfert résultant d'interactions chimiques fortes, ou non solvatant.
1.13 Principe de l’extraction liquide-liquide :
III.3. Principe de l’extraction liquide-liquide :
C'est une opération fondamentale de transfert de matière entre deux phases liquides non miscibles. L'extraction liquide-liquide parfois appelée extraction par solvant consiste à transférer un ou plusieurs solutés contenus dans une solution liquide Lo vers un autre liquide non miscible So (le solvant). La concentration finale du soluté dans chaque phase (L et S) dépend de sa solubilité dans les liquides mis en jeu. Le solvant s'enrichi en soluté et est alors appelé extrait (E), la solution de départ s'appauvrit en soluté, elle est alors appelée raffinat (R). La solution initiale Lo et le solvant pur So sont mis en contact pour favoriser le transfert du soluté. Généralement, la densité des deux phases liquides sortantes sont différentes et peuvent donc être séparées par décantation. Ces deux étapes sont réalisées dans un bac mélangeur-décanteur représenté sur la figure III.1.
Ainsi l'extraction liquide-liquide est une opération de séparation qui fait intervenir le transfert de matière d'un soluté entre deux phases liquides totalement ou partiellement miscibles entre elles.
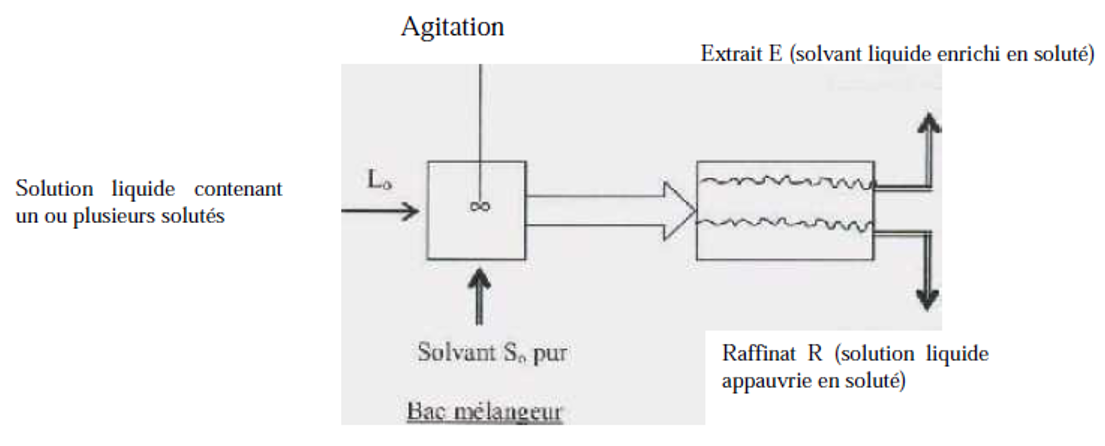
Figure III.1 : Schéma d'un bac mélangeur décanteur.
Contrairement à la distillation, le soluté ne change pas d'état au cours du transfert. La thermique intervient essentiellement en modifiant la solubilité du soluté dans chaque phase.
Pour que l’opération soit réalisable il est nécessaire :
- que les deux phases ne soient pas complètement miscibles
- que leurs masses volumiques soient différentes
- qu’il n’existe pas de réactions chimiques entre les divers constituants du mélange.
1.14 Différents types d’extraction
III.4. Différents types d’extraction
III.4.1. Extraction simple
On cherche à extraire un soluté contenu dans un mélange liquide homogène constitué de soluté et de diluant, nommé charge. On utilise pour cela une certaine quantité d'un solvant qui ne doit pas être miscible avec le diluant, mais être miscible avec le soluté.
Les deux phases liquides charge + solvant sont mélangées intimement (mélange non homogène) et le soluté se distribue dans chacune des phases selon un équilibre physicochimique.
Le mélange non homogène est alors décanté en deux phases non miscibles (Figure II.2):
- · l'extrait, contenant majoritairement du solvant et le soluté extrait,
- · le raffinat contenant majoritairement du diluant, ainsi que le soluté non extrait.
Figure III.2 : Schéma de principe d’une extraction simple.
III.4.2. Extraction multiple
III.4.2.1. Extraction à courant croisés
Dans une extraction à courant croisés (Figure II.3), ici à 3 étages, le solvant est (équitablement) réparti pour réaliser 3 extractions successives, la première sur la charge, la seconde sur le premier raffinat, la troisième sur le second raffinat.
On obtient en fin d'opération un raffinat (ici R3) et 3 extraits à 3 compositions différentes, le premier étant le plus concentré en soluté, le dernier le moins concentré. Les extraits sont mélangés entre eux dans la majorité des cas. Cette méthode donne un meilleur rendement qu'une extraction à simple étage pour une même quantité de solvant S, ici égale à S1+S2+S3.
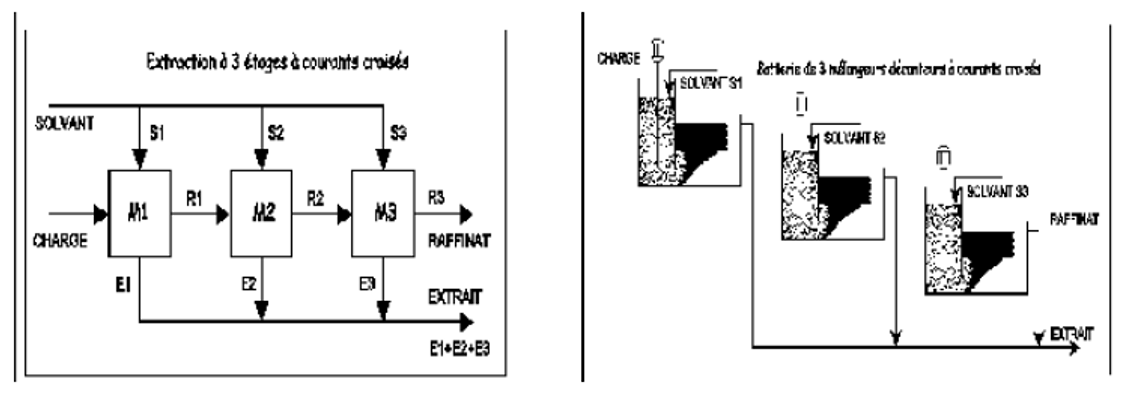
Figure III.3 : Schéma de principe d’une extraction à étages multiples.
1.15 Extraction à contre-courant
III.4.2.2. Extraction à contre-courant
Dans une extraction à contre-courant (Figure II.4), ici à 3 étages, on réalise 3 extractions successives en utilisant comme solvant l'extrait de l'étage suivant. On obtient en fin d'opération un raffinat (ici R3 sortant de l'étage n°3)) et un extrait (ici E1 sortant de l'étage n°1). On démontrera par des travaux dirigés que cette méthode donne un meilleur rendement qu'une extraction à simple étage ou à courant croisé avec le même nombre d'étages et la même quantité de solvant.
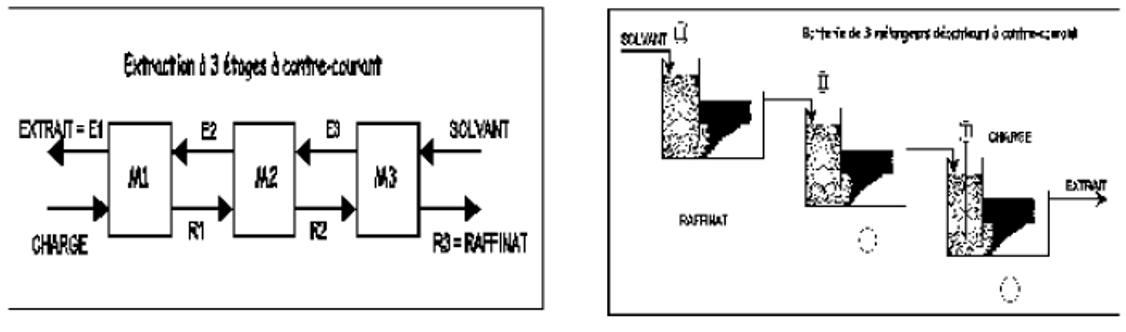
Figure III.4 : Schéma de principe d’une extraction à étages multiples à contre-courant.
III.5. Coefficient de partage ou de distribution
La distribution, ou le partage d’un soluté entre les deux phases a l’équilibre est donnée par le coefficient de partage (ou de distribution, ou de répartition). Cette grandeur se définit comme le rapport des teneurs respectives en soluté dans l’extrait et le raffinat lorsque l’équilibre est réalisé.
Chaque équilibre y = f(x) est représenté par une courbe appelée courbe de partage et un coefficient de partage :
Kc = yA/xA (II-1)
yA et xA sont les titres en soluté dans l’extrait et le raffinat respectivement.
III.6. Facteurs caractérisant la séparation par solvant
Ils dépendent de l'équilibre thermodynamique entre les phases. Le solvant doit non seulement permettre la séparation des produits, mais aussi être utilisable aisément dans les extracteurs et son emploi doit être aussi économique que possible. Dans ce but, il est nécessaire que le solvant réponde à un certain nombre de spécifications, parfois difficilement compatibles. Le solvant doit :
- bien solvater le soluté (mieux que le diluant)
- être bon marché
- ne pas être (trop) toxique/corrosif/polluant…
- être recyclable
- être de densité différente de celle du diluant et peu miscible avec lui …
1.16 Sélectivité du solvant
III.6.1. Sélectivité du solvant
Elle traduit la facilité que possède ce dernier à dissoudre un constituant de la charge préférentiellement à un autre. Le coefficient de sélectivité β analogue au coefficient de volatilité relative i utilisé en distillation exprime cette sélectivité. Avec les notations :
La séparation est d'autant plus aisée que β est plus grand, β égale 1 au point critique et pour l'azéotrope.
III.6.2. Pouvoir solvant
Il exprime la quantité de produits que peut dissoudre le solvant, tout en gardant un β acceptable. Si le pouvoir solvant est faible, il faut utiliser une grande quantité de solvant pour effectuer une séparation donnée.
III.6.3. Différence de masse volumique (Δρ) entre les solutions en contact
Une grande différence de masse volumique favorise la décantation des solutions et par suite augmente la capacité des décanteurs. Il est évident que les phases en contact (sensiblement en équilibre dans un décanteur, jamais en équilibre dans une colonne à remplissage) ne doivent à aucun moment atteindre la même densité.
III.6.4. Viscosité
Un solvant de faible viscosité doit être choisi, si possible, afin d'éviter les entraînements de gouttelettes et d'obtenir de hauts débits de fonctionnement, c'est-à-dire une capacité élevée de l'extracteur.
III.6.5. Tension interfaciale
Une faible tension interfaciale favorise la réalisation d'une grande surface de contact entre les phases, mais gêne la décantation de l'émulsion.
III.6.6. Facteurs économiques
Le solvant ne doit pas être: coûteux, toxique, corrosif, instable. Il doit posséder un haut point d'inflammabilité et un bas point de congélation pour être employé facilement. Il est nécessaire de pouvoir récupérer aisément le solvant contenu dans les solutions traitées, cette récupération étant ordinairement un des points économiquement importants du procédé.
Le procédé sera d'autant plus économique qu'il permettra de traiter des charges différentes avec le même solvant (souplesse de traitement).
III.7. Applications industrielles
L'essor industriel des procédés d'extraction liquide-liquide en chimie minérale a son origine dans les besoins de l'industrie nucléaire en sels de pureté exceptionnelle. Au cours de la période 1960-1970 on a assisté à l'application généralisée de cette technique à l'hydrométallurgie, favorisée par la mise.au point d'un complexant spécifique du cuivre (chélate). Actuellement fonctionnent 200 unités industrielles hydro métallurgiques, assurant la récupération, la séparation et la purification de très nombreux métaux.
De plus l'extraction liquide-liquide est appliquée à la fabrication de l'acide phosphorique très pur, du brome, du nitrate de potassium, et des acides nitrofluorydriques.
En chimie organique, les applications sont aussi nombreuses et importantes tant du point de vue quantitatif (pétrochimie) que qualitatif (industries alimentaires et pharmaceutique, récupération des polluants dans des effluents d'usine).
Actuellement, ce procédé de séparation des constituants d'un mélange et/ou de concentration est tout particulièrement utilisé lorsque les conditions technologiques ou physico-chimiques lui sont favorables, comme c'est le cas pour :
- La séparation des composés à température d'ébullition voisine (séparation de certains hydrocarbures aromatiques et aliphatiques).
- La séparation d'azéotropes eau-acides minéraux.
- La séparation de composés thermosensibles ou instables (obtention des antibiotiques).
- La concentration et la purification de solutions diluées, opérations souvent plus économiques que la distillation (cas des solutions diluées de sels métalliques tels que cuivre, uranium, vanadium).
- La substitution à la cristallisation fractionnée (séparation tantale- niobium).
- La séparation d'éléments ayant des propriétés chimiques voisines (séparation uraniumvanadium et hafnium-zirconium).
- L'obtention de produits de haute pureté (sels d'uranium de pureté nucléaire, sels de terres rares destinés à l'industrie optique ou électronique) ;
- Des séparations devant être effectuées de manière automatisée dans un environnement hostile (traitements des combustibles nucléaires irradiés).
1.17 Données d’équilibre liquide-liquide
IV.1. Règle des phases
L’extraction liquide-liquide est régie par une loi très importante due à W. Gibbs (1878) et que l’on connait sous le nom de règle des phases.
a- Phase
Des masses distinctes mais de composition chimique et d’états physiques identiques forment une seule phase, c’est le cas du mélange benzène et toluène. Donc c’est un ensemble homogène ayant les mêmes propriétés physico-chimiques et séparé des autres parties du système par une surface définie.
b- Système
C’est un corps ou un groupe de corps en interaction, le système peut être homogène ou bien hétérogène.
c- Constituants
Ce sont les substances chimiques qui composent un système, cas du benzène et du toluène qui forment une seule phase en étant deux constituants du système.
d- Variance
La variance V d’un système est le nombre de paramètres intensifs et indépendants qui caractérisent un état d'équilibre. D'une autre manière, c'est le nombre maximum de paramètres que peut fixer librement l'expérimentateur sans rompre l'équilibre.
Plaçons un liquide dans un récipient où l’on a préalablement fait le vide. Une certaine quantité de liquide se transforme aussitôt en vapeur et occupe la partie du récipient qui n’est pas remplie de liquide. On dit alors que le liquide est en équilibre avec sa vapeur. Il y a équilibre tant que les trois facteurs température, pression et la composition du liquide ainsi que sa vapeur ne varieront pas. La règle des phases appelée aussi loi d’équilibre de phase est fondée sur le deuxième principe de la thermodynamique et est exprimée par :
Φ + V= n+2 (III-1)
Où Φ ; est le nombre de phases / V : la variance n : le nombre de constituants
IV.2. Représentation des équilibres
Dans un système d’extraction liquide-liquide trois cas se présentent :
a- Les deux phases sont totalement miscibles
Ce cas est théorique, les deux phases raffinat et extrait sont totalement miscibles, donc il existe une seule phase et par suite aucune extraction du soluté n’est possible.
b- Les deux phases ne sont pas miscibles
Le soluté se répartit entre les deux phases, les valeurs de l’équilibre sont données par les rapports des : poids/raffinat et soluté/extrait.
c- Les deux phases sont partiellement miscibles
Donc le problème est plus compliqué, on fait recours à une représentation graphique (diagramme ternaire) permettant d’exprimer les solubilités réciproques des trois constituants (raffinat, extrait et soluté) à l’état d’équilibre.
IV.3. Système ternaire (diagramme triangulaire)
IV.3.1. Equilibre liquide-liquide d'un système ternaire
Les équilibres liquide-liquide ternaires mettent en jeu trois constituants : diluant (B), soluté (A) et solvant (S). Comme il a été mentionné dans le deuxième chapitre, l'introduction du solvant dans le mélange soluté-diluant provoque l'apparition de deux phases liquides, la première riche en diluant qu'on appelle raffinat, la seconde riche en solvant dite extrait.
IV.3.1.1. Représentation et lecture des diagrammes ternaires (triangulaires)
La présence des trois constituants dans le mélange liquide, le soluté A et les deux constituants B et S, nous conduit à adopter une présentation graphique au moyen des triangles équilatéraux.
Chaque sommet représente un constituant pur A, B ou S. Sur chaque côté du triangle on peut indiquer la composition des mélanges binaires (entre 0 % et 100 %) A-B, B- S, A- S.
Chaque point au cœur du triangle représente un mélange ternaire A-B- S. Il s'agit donc de positionner le point représentatif en fonction de la proportion de chaque constituant sachant que :
% A + % B + S % = 100 %. (III-2)
La figure (III.1a) donne les règles de lecture du diagramme.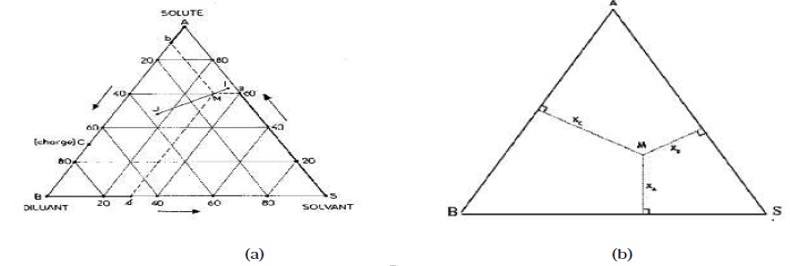
Figure IV.1 : Principe de lecture des compositions sur les diagrammes ternaires.
Cette représentation repose sur la propriété suivante des triangles équilatéraux: La somme des segments découpés sur les côtés par des parallèles à ceux-ci, à partir d'un point du triangle, est égale à un côté (Figure IV.1b) où :
(IV-3)
On établit facilement que le mélange des solutions I et J donne une solution globale représentée par un point M situé sur IJ et tel que :
(IV-4)
Les coordonnées des points I et J précisent les compositions des solutions correspondantes, les lettres I et J représentent leurs quantités (en poids si les concentrations sont en % poids par exemple).
Il se peut que le point M n'ait pas de signification physique, c'est-à-dire qu'il ne lui corresponde pas une phase homogène mais au contraire un extrait E et un raffinât R en équilibre (Figure III.2). D'après la règle des moments chimiques, on aura:
(IV-5) et M R E seront alignés; c'est une ligne d'équilibre.
Dans le cas de la figure III.3 les extraits et les raffinats tendent l'un vers l'autre quand leur teneur en soluté A croît; les lignes d'équilibre raccourcissent pour se transformer en un point critique (Pc).
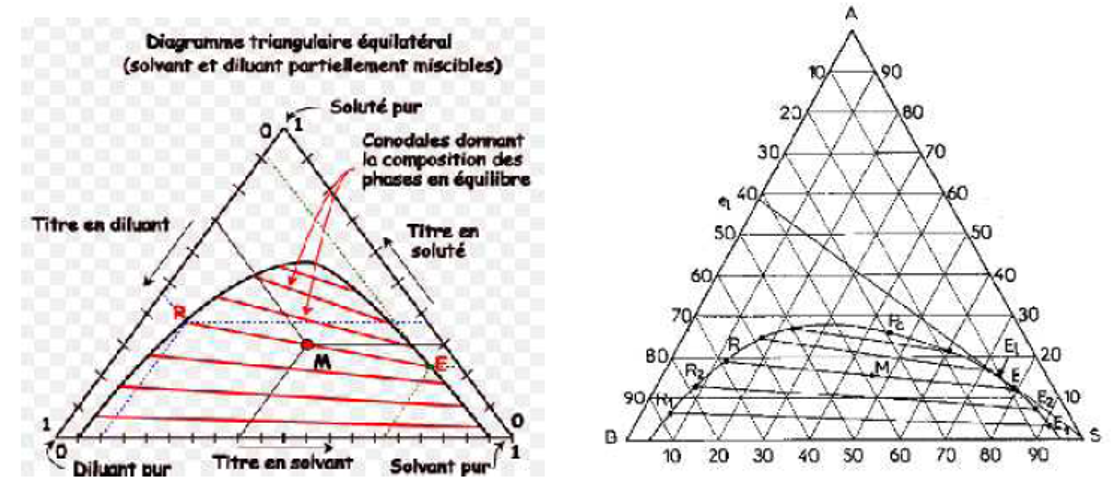
Figure IV.2: Représentation triangulaire équilatérale. Figure IV.3: Isotherme de solubilité
La courbe, lieu des solutions saturées, s'appelle l'isotherme de solubilité; elle divise le triangle en plusieurs régions dont l'une correspond à des mélanges globaux M se résolvant en 2 phases en équilibre (zone d'immiscibilité). Quand une ligne est horizontale, il y a solutropie; l'azéotropie correspond au cas où la ligne d'équilibre passe par le point S.
1.18 Règle des mélanges: relation barycentrique
IV.3.1.2. Règle des mélanges: relation barycentrique
Considérons un mélange ternaire (A + B + S) de masse R kg qui est mélangé à un mélange ternaire (A + B + S) de masse E kg mais de composition distincte. Le point M représentatif du ternaire (A + B + S) final a une composition intermédiaire entre R et E qui se situe graphiquement en un point de la droite RE dont on peut déterminer la position en écrivant les bilans massiques à l'aide de la figure IV.4.
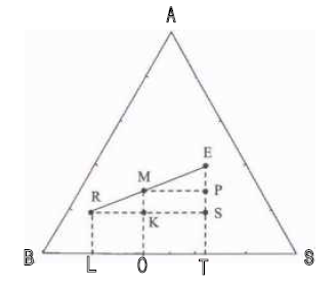
Figure IV.4: Relation barycentrique entre les compositions de M, R et E.
R+E = M (IV-6)
R : masse (kg) du mélange R
E : masse (kg) du mélange E
M : masse (kg) du mélange M
• Bilan massique sur le constituant C.
Soit :
(IV-7)
Les relations dans les triangles semblables donnent :
(IV-8)
(IV-7) + (IV-8) conduit à la règle des phases ou règle des segments inverses :
(IV-9)
Cette relation permet de positionner le point M à partir des masses R et E.
Ainsi lorsque : le point M est plus proche de R que de E.
Si : le point M est plus proche de E que de R.
Les bilans massiques sur les trois constituants conduisent aux relations qui traduisent le fait que le point M est le barycentre des points R et E, affectés de coefficients égaux aux masses totales des mélanges correspondants.
1.19 Expression des propriétés physico-chimiques
Si l'on représente à pression et température fixées un système dont deux constituants (et deux seulement) ne sont pas miscibles, on obtient le plus souvent un diagramme du type de celui de la figure IV.5.
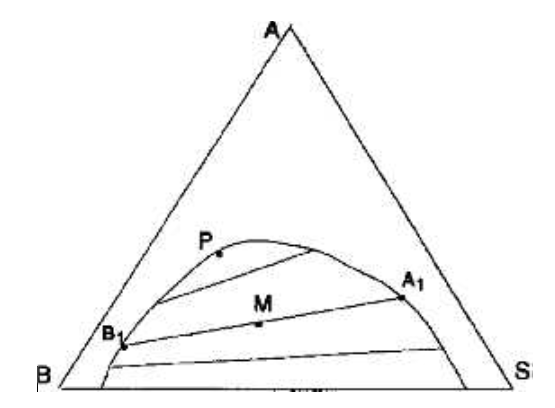
Figure IV.5: Diagramme triangulaire contenant une courbe binodale.
La figure IV.5 représente l'allure du système éthanol (A), eau (B) et benzène (S) à la température ambiante. La courbe binodale sépare le domaine à une phase en un domaine à deux phases. Les phases saturées, en équilibre, se trouvent aux extrémités de segments qui sont les droites de conjugaison. Le faisceau des droites de conjugaison commence par un segment appartenant au mélange binaire BS; il s'achève sur un point limite (P), qui correspond à une droite de conjugaison tangente à la courbe binodale. Les droites de conjugaison, comme la courbe binodale doivent être déterminées expérimentalement. Par contre, quand le diagramme est connu, les propriétés barycentriques permettent quelques prévisions.
De façon à alléger ce diagramme, on peut construire, à l'aide des droites de conjugaison connues, une courbe conjuguée par rappel parallèlement aux côtés du triangle. Cette courbe tracée, on peut ne laisser subsister qu'elle sur le diagramme et construire avec son aide les seules droites de conjugaison, utiles (Figure IV.6).
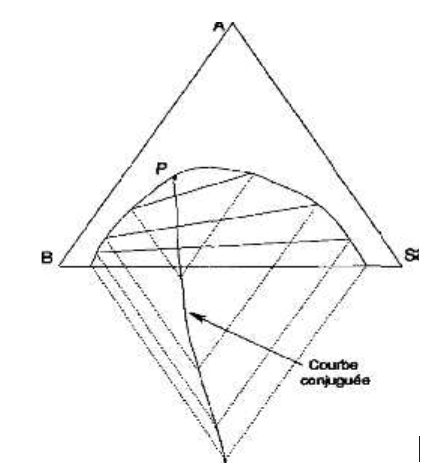
Figure IV.6: Tracé des droites de conjugaison.
Des différences plus radicales s'observent dans les systèmes qui présentent deux paires de liquides non totalement miscibles. On observe souvent des systèmes comme celui de la figure
IV.7 qui illustre le ternaire : méthyléthylcétone (A) eau (B) et chlorobenzène (S).
Figure IV.7: Deux paires de liquides non miscibles une seule lacune de miscibilité.
Des systèmes comme celui de la figure IV.7 sont engendrés quand les lacunes de miscibilité des deux binaires (AB) et (BS) croissent quand la température varie (dans le sens de l'augmentation quand les mélanges sont endothermiques) et qu'à la température où les deux lacunes deviennent contiguës, les deux points limites coïncident (Figure IV.8).
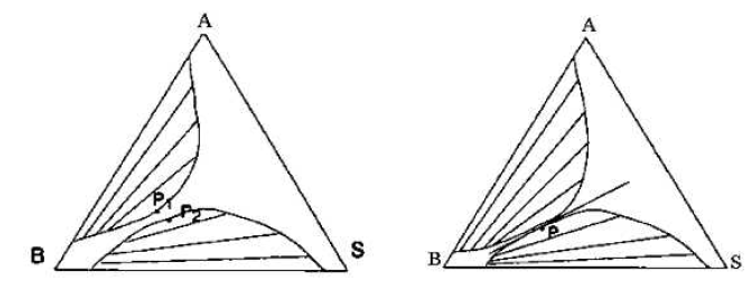
Figure IV.8: Contact des lacunes des miscibilités avec fusion des points limites.
Au contraire, quand les points limites ne fusionnent pas, on obtient un domaine à trois phases liquides: voir la figure IV.9. Quand ce domaine à trois phases existe, on remarque qu'il existe trois domaines diphasiques différents (au lieu de deux dans le cas contraire).
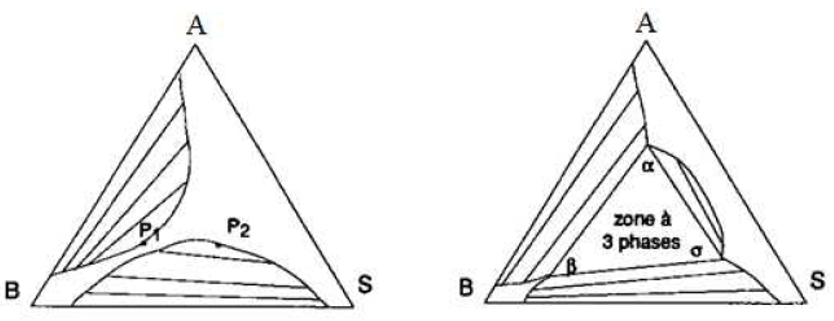
Figure IV. 9: Apparition d'un domaine à trois phases liquides.
Le domaine triphasique est un triangle dont les sommets sont α, β et σ. À la température considérée, tout mélange dont la composition est telle que son point représentatif se situe dans ce triangle se sépare en trois mélanges de composition α, β et σ, selon les règles barycentriques en effet, la variance est nulle et toutes les compositions sont déterminées.
Notons enfin que la présence de solides, que l'on doit considérer dès que l'on s'intéresse aux partages de sels métalliques entre deux phases liquides, conduit à des diagrammes très variés: nous donnons à titre d'exemple l'allure des diagrammes éthanol, eau et fluorure de potassium d'une part et eau, acétone et soude d'autre part sur la figure IV.10.
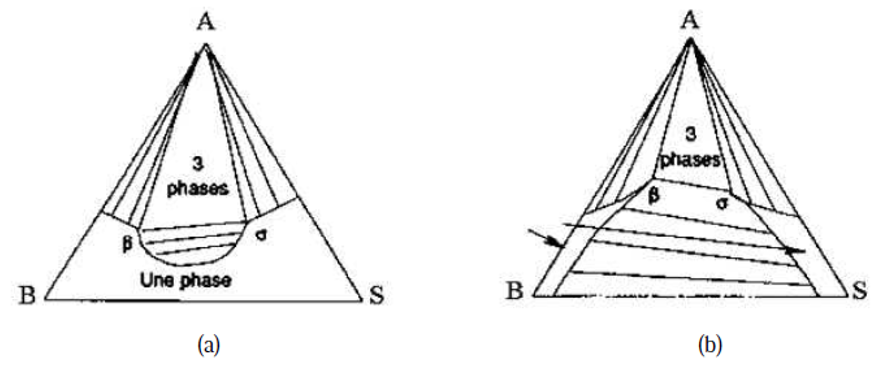
Figure IV.10: Diagramme avec un solide : (a) le soluté est le KF, (b) le soluté est NaOH.
1.20 Système ternaire: Autres diagrammes
IV.3.2. Système ternaire: Autres diagrammes
IV.3.2.1. Diagramme rectangulaire
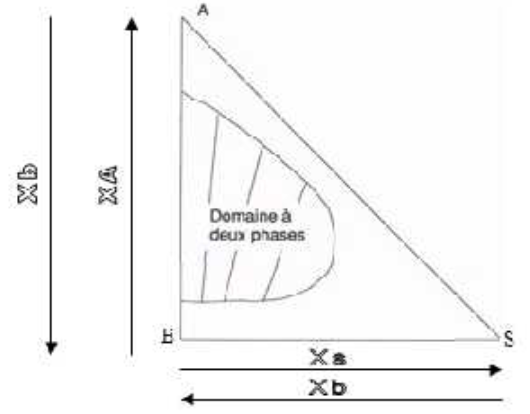
Figure IV.11: Diagramme triangle rectangle.
Ni la physico-chimie, ni les propriétés barycentriques n'imposent que le triangle que l'on utilise comme support du diagramme d'équilibre liquide-liquide soit un triangle équilatéral.
L'emploi de rappels à 60° demande un matériel adapté et est plus contraignant que celui de rappels orthogonaux. C'est pour cette raison qu'à des fins appliquées, on utilise le triangle rectangle en plus de l'équilatéral.
La figure IV.11 donne un exemple de diagramme établi sur un triangle rectangle, qui met en évidence comment le problème de la lecture des coordonnées y est résolu.
IV.3.2.2. Diagramme de Janecke
On utilise également un autre diagramme, qui n'est pas un diagramme triangulaire. C'est un diagramme qui comprend deux axes orthogonaux, sur lesquels on porte des cordonnées dites
«de Janecke», qui sont définies de la façon suivante. Si un mélange M contient les trois constituants A, B et S, en notant les masses de ces constituants par les mêmes lettres A, B et S, il vient :
(IV-10)
On constate immédiatement que les coordonnées de ce diagramme qui est représenté sur la figure IV.12, jouent des rôles dissymétriques: X varie entre 0 et 1, tandis que Y varie entre 0 et l'infini. En effet, le solvant pur S est un point à l'infini.
Pour utiliser le diagramme de Janecke, il est indispensable, connaissant les points représentatifs de deux mélanges P et Q, de savoir placer le point représentatif de M = P + Q.
Le diagramme de Janecke, malgré le caractère apparemment artificiel des coordonnées qu'il emploie, présente à un degré supérieur les mêmes qualités que le diagramme triangle rectangle. On peut même le considérer comme un cas extrême de triangle rectangle dont l'un des sommets aurait été rejeté à l'infini.

Figure IV.12: Diagramme de Janecke.
IV.3.3. Systèmes ternaires: Diagrammes qui ne décrivent pas totalement les équilibres.
IV.3.3.1. Diagramme de distribution
C'est un diagramme qui permet de représenter de façon immédiate la distribution (ou partage) du constituant à extraire entre les deux phases liquides en équilibre. On porte en abscisse la fraction massique de A dans les phases riches en B, soit xa(p) que nous noterons x et en ordonnée la fraction massique de A dans les phases riches en S, xa(q) que nous noterons y.
Chaque point de la courbe représente un équilibre et se déduit de la position des droites de conjugaison sur le diagramme triangulaire.
La courbe de partage a deux contacts avec la première bissectrice, le point limite PI, qui correspond au point où les deux branches de la courbe binodale se rejoignent, et l'origine. La courbe de partage passe par l'origine, qui correspond à l'équilibre binaire entre B et S. Comme toute courbe ayant une origine expérimentale, la courbe de partage admet une tangente à l'origine: en conséquence, le rapport Da = y/x, connu sous le nom de coefficient de distribution, tend vers une valeur constante quand xa tend vers zéro. Cette propriété se conserve quel que soit le système de coordonnées, en particulier quand on l'exprime avec des concentrations au lieu de fractions massiques (ou molaires).
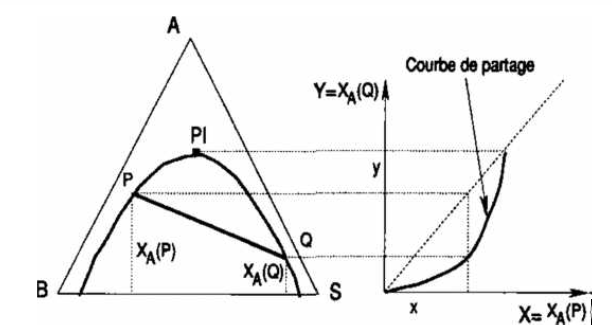
Figure IV.13: Construction de la courbe de partage.
On peut noter dès maintenant, car nous utiliserons cette propriété plus loin, que tout faisceau de droites qui coupent la courbe binodale sur le diagramme triangulaire définit une courbe sur le diagramme de distribution (Figure IV.13).
1.21 Diagramme de sélectivité
IV.3.3.2. Diagramme de sélectivité
Etant donné deux mélanges en équilibre, P riche en B et Q riche en S, ce diagramme s'obtient en portant respectivement les quantités suivantes en abscisse et en ordonnée :
Ces coordonnées ne sont autres que les abscisses du diagramme de Janecke. On construit donc le diagramme de sélectivité à partir du diagramme de Janecke tout comme on construit le diagramme de distribution à partir d'un diagramme triangulaire. Ces coordonnées, à la différence des fractions massiques ne contiennent pas le solvant S et permettent donc d'estimer effectivement la sélectivité b du solvant, c'est-à-dire son aptitude non pas à extraire A, mais à l'extraire mieux que B. b est défini de la façon suivante :
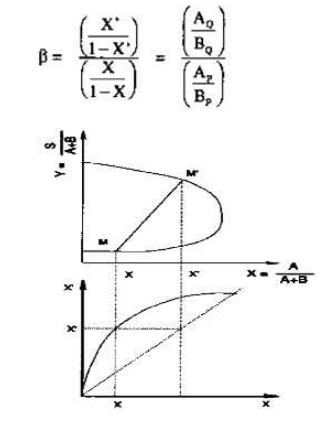
Figure IV.14: Construction de la courbe de sélectivité.
Sur la figure IV.14, on peut noter que la courbe de partage se trouve tout entière au-dessus de la première bissectrice, ce qui correspond au fait que le constituant privilégié A se partage préférentiellement du côté du solvant S entre B et S.
IV.4. Influence de la température
En général une diminution de la température de traitement se traduit par une diminution de la zone de miscibilité sur le diagramme ternaire (voir figure IV.15a).
II faut remarquer aussi que la température a une influence sur la pente des lignes d'équilibre. Ainsi, pour pouvoir évaluer le degré de séparation à différentes températures, faut-il se reporter aux courbes de sélectivité (Figure IV.15b). En général le degré de séparation augmente quand la température diminue.

Figure 15a: Influence de la température sur Figure 15b: Influence de la température
la zone de miscibilité sur les courbes de sélectivité