Chapitre 3 : Réactions biologiques faisant intervenir des métaux
Chapitre 3 : Réactions biologiques faisant intervenir des métaux
| Site: | Touch By SukaJanda01 |
| Cours: | CHIMI 5121 : Chimie bio inorganique |
| Livre: | Chapitre 3 : Réactions biologiques faisant intervenir des métaux |
| Imprimé par: | Visiteur anonyme |
| Date: | mardi 17 juin 2025, 13:33 |
1 Réactions d’échanges électroniques ou d’oxydo-réduction
a) Généralités
Oxydo-réduction : échange irréversible d’électrons entre une espèce donnant un électron (Réducteur, Red1) et une espèce captant cet électron (Oxydant, Ox2).
L’espèce qui donne l’électron est oxydée. Celle qui capte l’électron est réduite.
Oxydation: perte d’électron
Red1 génère la forme oxydée (Ox1). Ox2 génère la forme réduite (Red2).
Réduction: gain d’électron
Oxydo-réduction : échange d’électrons entre les couples rédox Ox1/Red1 et Ox2/Red2.
b) Degré d’oxydation
Caractérise l’état d’oxydation de l’élément.
N’est pas une charge réelle, mais un état fictif, un outil conventionnel qui caractérise la configuration électronique de l’élément et ses propriétés.
c) Prévision des réactions rédox
Réaction précédente permise si : E°(Ox2/Red2) > E°(Ox1/Red1)
Un oxydant réagit avec un réducteur de potentiel normal inférieur au sien :
d) Echelle de potentiels normaux et conséquences
Cuivre: 0 (Cu, métallique) ; + I (Cu+, cuivreux) ; + II (Cu2+, cuivrique). Etats d’oxydation les plus usuels : 0 et + II.
Cu+ : beaucoup moins usuel, instable en solution aqueuse.
E° (Cu+/Cu) = + 0,52 Volt
E° (Cu2+/Cu+) = + 0,17 Volt
E° (Cu2+/Cu) = + 0,35 Volt
En présence d’ammoniaque, espèces en présence : Cu(+II)(NH3)42+, Cu(+I)(NH3)2+ et Cu(0).
Cu+ + 2 NH3 → Cu+I(NH3)2+
Cu2+ + 4 NH3 → Cu+II(NH3)42+
Cu+II(NH3)42+ + e- → Cu+I(NH3)2+ + 2NH3 E°(Cu+II/Cu+I) = + 0,06 V
Cu+I(NH3)2+ + e- → Cu + 2NH3 E°(Cu+I/Cu) = - 0,13 V
Etat d’oxydation + I du Cuivre stable en présence d’ammoniaque :
Cu + Cu+II(NH3)42+ → 2 Cu+I(NH3)2+
Etat d’oxydation non usuel du Cuivre en solution aqueuse stabilisé en présence d’ammoniaque, sous forme d’un complexe « ammine ».
2 Exemples de réactions d’oxydo-réduction en milieux biologiques
2-1) Les fonctions métaboliques essentielles du Cuivre
a) Généralités
Le Cuivre : co-facteur clé dans de nombreux processus oxydo-réducteurs et pour le transport d’oxygène. En milieu biologique, Cuivre (+ I) et (+ II).
Cuivre + I : préférentiellement Cystéine et Méthionine (ligand S)
Géométrie tétraédrique (4 ligands) ou trigonale (3 ligands).
Cuivre (+ II) : préférentiellement ligands azotés (Histidine) ou oxygénés (Sérine, Thréonine, Tyrosine ou H2O)
Géométries variées (carré plan, bipyramide à base triangulaire, bipyramide à base carrée, …).
Les sites actifs répartis en 3 classes, en fonction de leurs caractéristiques spectroscopiques :
Type 1 (T1) : forte absorption vers 600 nm : coloration bleue intense de l’enzyme, d’où le nom de protéine cuivre bleue.
1 noyau métallique contenu dans le site actif.
→ bipyramide à base triangulaire ou tétraèdre déformés.
Type 2 (T2) : 1 centre métallique est incolore.
Principal ligand : l’Histidine.
Géométrie: moins restrictive plus grand nombre de structures.
Type 3 (T3) : 2 noyaux métalliques liés l’un à l’autre via un ligand
Forte absorption à 330 nm.
4ème classe : 3 noyaux métalliques sous forme de cluster à 3 centres : 1 centre (1 noyau) T2 et l’autre centre (2 noyaux) T3.
Exemple : Laccases, site T2/T3 (+ 1 troisième site métallique de type T1).
b) Galactose oxydase (EC 1.1.3.9)
α – Structure de la biomolécule
La Galactose oxydase : - enzyme extra-cellulaire
- un seul fragment polypeptidique (639 acides aminés)
- secrétée par certaines variétés de champignons et de moisissures pathogènes
β – Structure du métallobiosite
1 seul centre métallique, de type T2, au degré d’oxydation + II, à proximité de la surface de la protéine.
Pyramide à base carrée.
Cu entouré de 5 ligands :
2 ligands Histidines : His496 et His581 ;
2 ligands Tyrosines : Tyr272 et Tyr495 (en position axiale) ;
1 molécule d’eau H2O ou un acétate.
Galactose oxydase : seule métalloenzyme à posséder deux résidus Tyrosines dans son environnement de coordination.
Présence d’une liaison thioéther entre Tyr272 et Cys228, en position ortho de la fonction phénol du résidu Tyrosine.
C-S-C : liaison intramoléculaire formée par l’enzyme elle-même (« self-processing »), de façon réversible en présence d’oxygène O2 avec intervention du Cuivre, absolument indispensable à l’activité catalytique de l’enzyme.
Enzyme monomérique avec 2 sites oxydo-réducteurs.
γ - Fonction biologique de la Galactose oxydase
Catalyse la réaction d’oxydation d’alcools primaires en aldéhydes correspondant, notamment les dérivés de type galactose.
Réaction extrêmement sélective : systématiquement en position 6 du fragment galactose.
Réaction concomitante : réduction d’oxygène O2 en peroxyde d’hydrogène H2O2 : phénomène relativement rapide, pour la production rapide de peroxyde d’hydrogène afin d’assurer la défense de l’organisme contre les bactéries.
Réaction globale d’oxydation d’alcool primaire en aldéhyde :
R-CH2OH + O2 → R-CHO + H2O2
Alcool primaire aldéhyde
δ - Mécanisme
Grandes lignes connues mais mécanisme global non encore totalement élucidé.
Plusieurs hypothèses mais une certitude : nécessité d’un seul centre métallique mais de 2 électrons.
Oxydation de Cys228 par de l’oxygène et du Cuivre (+ II)
→ liaison thioéther avec Tyr272 puis le radical libre phénoxy.
Tyr495 récupére le proton labile de la fonction alcool primaire du substrat.
L’alcoolate réagit ensuite avec le radical phénoxy Tyr272
→ radical cétyle extrêmement réactif
→ transfert d’un électron en excès du cétyle vers l’atome central Cuivre (+ II). Réduction du Cu (+ II à + I) et transformation du cétyle en aldéhyde.
En présence d’oxygène O2, Cuivre + I est réoxydé en Cuivre + II, et O2 est réduit en peroxyde d’hydrogène H2O2, en régénérant le site actif initial.
c) Tyrosinase (EC 1.14.18.1)
α – Structure de la biomolécule
Tyrosinase : différentes enzymes qui catalysent l’oxydation de noyaux phénoliques, comme la Tyrosine, ou polyphénoliques.
Tyrosinases : isolées à partir de nombreux organismes, très fréquentes parmi les eucaryotes, mais également chez certains procaryotes.
Toutes ces enzymes n’ont pas forcément la même structure :
Tyrosinases issues des Streptomyces genus, Streptomyces glaucescens et Neurospora crassa : monomériques.
Tyrosinase issue de la moisissure de l’Agaricus bisporus : protéine tétramérique, comportant deux types de sous-unités différentes.
Tyrosinase d’origine humaine : glycoprotéine.
β – Structure du métalobiosite des Tyrosinases
2 centres métalliques T3, chaque centre est coordiné par 3 ligands Histidines (N) (triangulaire). 1 molécule d’O2 peut s’intercaler entre ces deux centres.
γ – Fonction biologique des Tyrosinases
Tyrosinases : très fréquentes dans les moisissures et chez les vertébrés, catalysent l’étape initiale de la formation de la mélanine et autres pigments, à partir de la Tyrosine.
Elles catalysent, deux types de réactions, liées l’une à l’autre :
ortho-hydroxylation d’un mono-phénol, en ortho-diphénol ou catéchol :
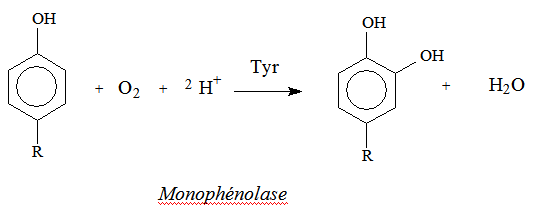
→ oxydation à deux électrons de l’ortho-diphénol en ortho-quinone.
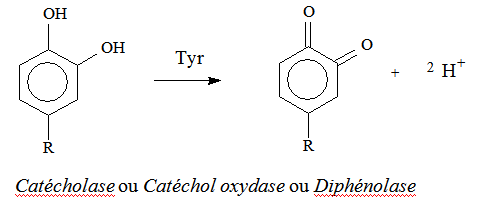
δ – Mécanisme
Oxygène incorporé dans le substrat lors de l’ortho-hydroxylation, provient d’O2.
Les deux électrons requis pour assurer la réduction du second atome d’oxygène en H2O proviennent du substrat
Les noyaux métalliques cuivriques conservent leur degré d’oxydation (+ II).
→ L’enzyme assure l’intégralité des transferts électroniques.
Première possibilité : - intercalation d’O2 entre les centres métalliques
- fixation d’un phénol, par l’un des centres cuivriques
- un oxygène intercalé réagit sur le noyau aromatique pour générer un précurseur du diphénol.
Deuxième possibilité, après fixation d’O2 entre les centres métalliques :
- fixation d’un noyau aromatique portant deux groupements hydroxy en position ortho
- Réaction avec l’oxygène intercalé
→ génération d’ortho-quinone et H2O.
- Présence d’un second pont hydroxy entre les deux centres métalliques
→ fixation d’une nouvelle molécule ortho-diphénol et répétition du processus.
d) Laccases (EC 1.10.3.2)
α – Structure de la biomolécule
Laccase : famille de glycoprotéines faisant partie des Multicopper Blue Proteins (MBP ou protéines bleues à plusieurs noyaux cuivres).
Classées en 2 groupes, en fonction de leur provenance (plantes ou moisissures de champignons).
Egalement présentes dans quelques bactéries.
Fort degré de similitude entre les Laccases, avec certaines différences locales.
β – Structure du métallobioiste des Laccases
Un trait commun des Laccases : 4 centres métalliques cuivre, 1 T1, ou Cuivre bleu, et un cluster à 3 noyaux T2/T3.
Cuivre T1 connecté au cluster trinucléaire T2/T3 par un fragment tripeptidique His-Cys-His.
T1 et T2/T3 : séparés de 12,5 Å, caractéristique des laccases.
γ – Fonction biologique des Laccases
Laccases : catalysent l’oxydation d’ortho- et para-diphénols, amino-phénols, polyphénols, polyamines, aryldiamines, et certains ions inorganiques.
Réaction d’oxydation couplée à une réaction de réduction de O2 en eau.
Exemple : oxydation des lignanes (phénols) qui en polymérisant en lignine, polyphénol, de masse molaire est supérieure 10 000 g.mol-1 (10 kDa).
δ – Mécanisme
Plusieurs hypothèses mécanistiques ont été proposées mais le substrat est systématiquement oxydé à proximité du site T1.
Les électrons libérés au cours de cette réaction sont transférés au cluster T2/T3.
Transfert électronique rendu possible du fait de la proximité des centres métalliques (distance : 12,5 Å).
Le substrat est oxydé à proximité du site T1 qui est réduit en Cu+.
Cet électron est ensuite transféré à un autre centre métallique cuivrique de type T2 ou de type
T3, régénérant le centre cuivrique T1 et un centre métallique cuivreux T2 ou T3.
Le noyau cuivrique T1 peut dès lors être impliqué dans un nouveau processus d’oxydation du substrat tout en étant à nouveau réduit en Cu+.
Ce processus se répète jusqu’à ce que tous les centres métalliques se retrouvent à l’état de Cu+ forme totalement réduite qui ne peut plus participer à l’oxydation d’un substrat.
Intervention d’O2 pour régénérer les centres cuivriques et reprise du processus réactionnel.
e) Quercétinase (EC 1.13.11.24)
α – Structure de la biomolécule
Quercétinase ou 2,3-QD : glycoprotéine homodimère (2 monomères protéiques identiques) de masse molaire voisine de 50 kDa avec deux noyaux Cuivre (1 Cu de type T2/monomère).
Exprimée par des champignons, se développant sur des milieux de culture contenant de la quercétine, comme source de Carbone et d’énergie.
β – Structure du métallobiosite
Centre métallique de type T2, entouré de 3 ligands Histidines : His66, 68 et 112.
2 géométries distinctes :
→ tétraèdre déformé : cuivre entouré de 3 ligands Histidines et 1 H2O
→ bipyramide trigonale : Glu73 participe à la complexation.
Les deux géométries sont importantes pour l’activité catalytique de l’enzyme.
γ – Fonction biologique
Quercétine : dérivé aromatique de la famille des flavonoïdes.
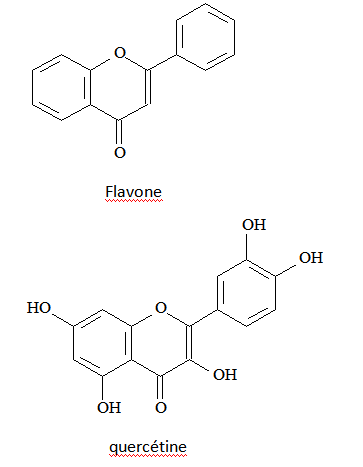
Flavonoïdes : molécules organiques présentes dans le monde végétal, assurant un grand nombre de fonctions biologiques.
Quercétine : un des flavonoïdes les plus abondants dans la Nature.
Quercétinase : catalyse la réaction de dioxygénation des flavonols comme la quercétine, en produisant du monoxyde de carbone et le depside correspondant (ester phénolique) :

→ consommation d’O2
→ piège à oxygène ou à radicaux libres.
δ – Mécanisme
1ère étape : génération d’une espèce activée :
- complexation du Cu(+II) par le substrat organique déprotoné par échange acidebase
avec Glu73 avec génération de Cu(+I)
→ activation du ligand organique, qui va réagir avec de l’oxygène moléculaire, en générant un intermédiaire bi-radical
→ formation d’un intermédiaire cyclique endo-peroxyde à 5 chaînons, dont 2 atomes d’oxygène liés entre eux
2ème étape : ouverture de ce cycle réactif par clivage de plusieurs liaisons covalentes, pour libérer le depside et CO.
2-2) Le Cobalt en milieux biologiques
a) Introduction
Cobalt : élément trace le moins abondant dans le corps humain : vitamine B12 et ses cofacteurs.
b) Vitamine B12 et co-facteurs
Existence: suggérée dans les années 1920 (Minot & Murphy, 1926).
Structure : élucidée par diffraction des rayons X (Hodgkin).
α – Structure de la métallobiomolécule
Vitamine B12, ou cobalamine.
Co : - degré d’oxydation + III
- géométrie octaédrique :
- entouré d’1 macrocycle tétraazoté (« corrine »)
- 2 ligands axiaux, dont un (CN) (ligand artificiel). Dans la Nature, « méthyle »
(méthylcobalamine) ou « adénosyle » (5-déoxyadénosylcobalamine)
Dans tous les cas, 1 trait commun : une liaison Co-C, ce qui en fait l’un des rares composés
organométalliques naturels.
β – Fonctions biologiques
Vitamine B12 et d’acide folique : impliqués dans la synthèse de l’ADN
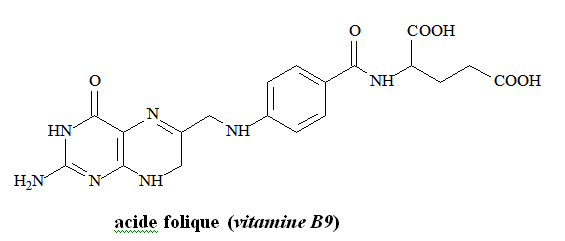
Vitamine B12 : co-facteur organométallique fondamental pour une réaction de transfert de groupement méthyle.
2 réactions enzymatiques dépendantes de la vitamine B12 sont connues :
- la conversion de l’Homocystéine en Méthionine.
- la conversion de l’acide méthylmalonique en acide succinique.
Déficience en vitamine B12 : dérèglement de ces processus biochimiques pouvant entraîner une hyper-homocystéinémie, i. e. augmentation du niveau d’Homocystéine dans le sérum sanguin.
γ – Mécanisme
La vitamine B12 et ses co-facteurs : rôle important dans différentes réactions, dont le transfert d’un méthyle vers l’Homocystéine pour générer la Méthionine.
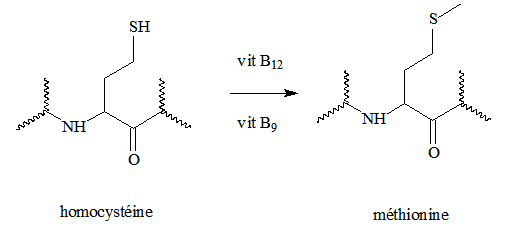
Réaction catalysée par la Méthionine synthétase, enzyme vitamine B12 dépendante.
Enzyme, de type Méthyl transférase, constituée d’une chaîne protéique de 1227 résidus acides aminés (masse molaire de l’ordre de 136 kDa).
Au cours de ce processus, la vitamine B12 accepte le groupement méthyle cédé par le CH3-H4 folate, avant de le transférer à l’Homocystéine. Le Cobalt est oxydé du DO + I au DO + III.
Les complexes de Cobalt sont des espèces très réactives, notamment au DO + I.
De nombreux mécanismes ont été suggérés. 3 proches de la réalité, sans certitude :
- mécanisme de type SN2, analogue à celui décrit en Chimie Organique
- addition oxydante d’un groupement méthyle sur l’atome de Cobalt
- transferts électroniques impliquant à chaque fois un seul électron
3 Autres exemples de réactions impliquant des métaux en milieux biologiques
a) Le Zinc en milieux biologiques
Zinc : requis pour l’activité de plus de 300 enzymes.
1ère enzyme Zn dépendante découverte en 1940 : l’Anhydrase carbonique II.
Métal essentiel pour la croissance, le développement et la différenciation de tout organisme vivant.
Métal trace le plus abondant dans l’organisme humain après le Fer.
Ion Zn2+ : une orbitale « d » pleine (3d10)
→ Zinc (+ II) très stable en milieu biologique oxydo-réducteur.
→ ne peut pas participer à un processus oxydo-réducteur.
Mais fonctionne comme un acide de Lewis en acceptant une paire électronique non liante.
- Zn2+ : énergie de coordination nulle, quelle que soit la géométrie du complexe
→ aucune géométrie n’est plus stable qu’une autre.
→ modification de la réactivité du son centre métallique en modifiant sa géométrie de coordination.
- Echange rapide de ligand du Zinc (+ II)
→ excellent partenaire catalytique permettant un « turn-over » efficace.
- Ligands préférés de Zn : S (Cystéine), N (Histidine) ou O (Aspartate, Glutamate).
Egalement : O (Tyrosine) et C=O (Asparagine, Glutamine).
Le centre métallique Zinc a deux rôles en milieu biologique :
ü rôle fonctionnel : participation à la réaction
ü rôle structural : participation au maintien et à la stabilisation de la structure tridimensionnelle de la protéine.
b) Anhydrase carbonique II (EC 4.2.1.1)
α – Structure de la biomolécule
Catalyse l’hydratation réversible du dioxyde de carbone :
CO2 + H2O → H+ + HCO3-
Se retrouve dans la plupart des organismes du règne animal, mais aussi des organismes participant à la photosynthèse, ainsi que dans certaines bactéries.
Absence d’homologie séquentielle significative entre les représentants des différentes classes d’Anhydrases carboniques.
Anhydrases carboniques : classées en 3 familles différentes : α, β et γ.
Anhydrases carboniques issues du règne animal sont de type α.
β – Structure du métallobiosite
Zinc : coordiné à 3 résidus Histidines : His94, His96 et His119
Géométrie tétraédrique déformée, complétée par une molécule d’eau.
γ – Fonction biologique
L’Anhydrase carbonique II, CA II, facilite l’échange de CO2 dans les érythrocytes, le foie et les poumons ; intervient dans l’homéostasie acido-basique et la sécrétion de HCO3-.
Chez les plantes, essentielle pour la fixation du CO2.
δ – Mécanisme
Initialement étudié à partir d’Anhydrase carbonique bovine et d’Anhydrases carboniques humaines I et II.
Les principales étapes du mécanisme réactionnel sont identiques quelle que soit l’enzyme impliquée dans ce processus.
1ère étape : attaque de la liaison Zn-OH par CO2 formation d’une liaison Zinc-Bicarbonate.
HCO3- est déplacé par de l’eau lors d’un échange de ligands.
Au cours de la seconde étape, l’ion H+ de cette molécule d’eau est transféré au solvant
(Tampon) par l’intermédiaire de l’His64, pour régénérer l’espèce catalytique active : le groupement Zn-OH.
Autre mécanisme : intervention du résidu Thr199.
Conclusion
La vie est essentiellement organique, mais, elle a évolué de façon à incorporer toutes les facettes de la chimie inorganique, susceptibles de lui être profitables.
13 éléments sont présents dans bon nombre d’organismes vivants.
4 sont présents en très fortes quantités.
Calcium : élément métallique le plus abondant de notre organisme. Une déficience en Calcium est responsable de la fragilité osseuse.
9 autres éléments : « éléments traces ».
Fer : élément métallique trace le plus abondant de notre organisme, dont la présence est vitale. Mais, le Fer : extrêmement toxique, si excès : hémochromatose.
Annexes
1 – La réaction de Belousov-Zhabotinsky
a – Généralités
b – Historique
Etude du mécanisme du cycle de Krebs en utilisant un modèle chimique inorganique.
Mais mise en évidence d’un système oxydo-réducteur complexe évoluant périodiquement :
« Réaction oscillante de Belousov-Zhabotinsky ».
c – Mécanisme réactionnel
Succession d’environ 80 étapes élémentaires et 26 variations de concentrations
BrO3- réduits par Br- en Br2.
Br2 réagit avec l’acide malonique pour générer l’acide bromomalonique.
L’acide bromomalonique réagit avec Ce(+IV) (jaune en solution, bleu en présence de ferroïne) en libérant des ions Br-.
Ce4+ réduit en Ce(+III) (incolore en solution, rouge en présence de ferroïne).
Les ions bromures réagissent sur BrO3- pour générer Br2 qui réagit sur l’acide malonique, pour redonner de l’acide bromomalonique ... jusqu’à disparition complète de Ce(+IV) solution rouge.
→ la concentration en ions Br- décroît.
A ce moment là, l’agent réducteur des bromates n’est plus Br-, mais HBrO2.
Les ions bromates sont réduits par HBrO2, en radicaux BrO2 qui sont eux-mêmes réduits en HBrO2, en présence des sels de Ce(+III) qui sont oxydés en Ce(+IV), changement de coloration de la solution du rouge au bleu.
L’acide bromomalonique peut à nouveau libérer des ions bromures en régénérant le Ce(+III).
Les ions bromures voient alors leur concentration augmenter progressivement. Au bout d’un certain temps, cette concentration est suffisante pour à nouveau réduire les ions bromates et ainsi recommencer le cycle des ions bromures.
d – Intérêts de la réaction de Belousov-Zhabotinsky ?
2 – La vitamine C, un phénomène oxydo-réducteur en milieu biologique
a – Généralités
La vitamine C : indispensable à notre organisme (propriétés anti-oxydantes et antiscorbutiques, entre autres).
L’homme : un des deux seuls mammifères incapables de synthétiser la vitamine C dont son organisme a besoin.
En théorie, la vitamine C ne devrait, en aucun cas, traverser les parois cellulaires.
Quelques minutes après son ingestion, elle se retrouve dans le compartiment cérébral.
b – Comment expliquer ce phénomène ?
Plusieurs hypothèses ont été proposées.
c – Quel est l’intérêt de ce phénomène ?