Méthodes de détermination de la géométrie d'un composé
1°) Le modèle de l’hybridation des orbitales atomiques
1-1 : Origine
1-2 : Principe de l’hybridation
1-3 : Hybridation sp
1-4: Hybridation sp2
1-5 : Hybridation sp3
1-6 : Hybridation sp3d
1-7 : Hybridation sp3d2
1-8 : Hybridation sp3d3
1-9 : Types d'hybridation et liaisons π
2°) Le modèle de la répulsion des charges ou modèle de Gillespie (VSEPR)
2-1 : Origine
2-2 : Principes de base
2-3 : Règles et notations VSEPR
2-4 : Cas des molécules avec liaisons multiples
2-5 : Cas des molécules ioniques
2-6: Effets du second ordre
2-7 : Limites de la méthode VSEPR
3°) Géométrie des complexes de coordination
4°) Géométries des complexes contenant des liaisons pontées
5°) Liaisons multi centrées
| Site: | Touch By SukaJanda01 |
| Cours: | Chimie minérale (Année 2015) |
| Livre: | Méthodes de détermination de la géométrie d'un composé |
| Imprimé par: | Visiteur anonyme |
| Date: | mardi 17 juin 2025, 02:08 |
1 Le modèle de l'hybridation des orbitales moléculaires
La théorie de l’hybridation a été développée au cours des années 1930, notamment par le chimiste américain Linus PAULING, prix NOBEL de Chimie en 1954. Elle permet de rendre compte de la géométrie de composés et de rendre aussi compte des liaisons présentes par le recouvrement d’orbitales. C'est une théorie descriptive de la liaison chimique qui eu, et a encore, un grand succès en chimie organique, car elle rend assez bien compte de faits expérimentaux, absolument incompréhensibles par la théorie de LEWIS, tels que l'existence de liaisons s et p.
Une liaison σ est obtenue par recouvrement axial de deux orbitales et une liaison π est obtenue par recouvrement latéral de deux orbitales. La modification de l’état des électrons externes des atomes concernés par la liaison, par rapport à l’état des atomes isolés est telle que nous parlerons de partage des électrons. Rappelons la configuration électronique de chaque atome isolé où l'électron n'est pas localisé au sens ponctuel du terme mais on peut décrire des régions de l'espace où cet électron a une forte probabilité de s’y trouver : c'est l'orbitale atomique. L’électron unique se trouve décrit par une orbitale atomique 1s. La stabilisation nous montre que lors du rapprochement de deux atomes, nous pouvons imaginer un recouvrement partiel de ces deux orbitales atomiques. Ce recouvrement est nécessaire pour assurer la plus grande stabilité correspondant à un recouvrement maximal, il ne se fera que suivant des directions privilégiées. Ceci permet d’interpréter le fait que la liaison covalente présente un caractère très fortement dirigé qui impose aux molécules des géométries bien caractéristiques que l’on repère par la mesure des angles entre les liaisons. Par exemple la molécule d’eau est coudée présentant un angle de 104,5° entre les deux liaisons O – H. Pour déterminer la géométrie d’une entité chimique, par cette théorie, on cherchera à déterminer l’état d’hybridation de l’élément central. Pour cela :
On commence d’abord par déterminer la structure de Lewis de cette entité chimique.
On compte ensuite le nombre de liaisons σ autour de l’élément central (on ne compte pas les π).
On considère ensuite la part de la structure électronique de l’élément central correspondant aux électrons de valence. Partant de cet état fondamental ainsi déterminé (donné directement par le tableau périodique), on passe à l’état excité de cet élément central en « cassant » tous les doublets de l’état fondamental par promotions électroniques des électrons vers les orbitales à énergies supérieures de façon à ce qu’il n’y ait qu’un seul électron par orbitale dans cet état excité.
On « sommes » ensuite autant d’orbitales dans l’état excité qu’il y a de liaisons σ autour de l’élément central en commençant par l’orbitale de plus basse énergie (c’est toujours une orbitale s dans les éléments non transitionnels). Ces n orbitales sommées vont donner naissance à n orbitales hybrides équivalentes contenant chacune un électron et ces orbitales hybrides, par recouvrement de type axial avec n’importe orbitale s ou p des éléments liés à l’élément central forment des liaisons σ. Un recouvrement de type latéral entre deux orbitales parallèles donne une liaison π.
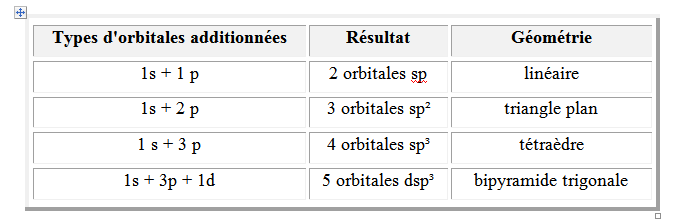
L’état d’hybridation est donné par les orbitales « sommées ». Il est de la forme spndm (n = 1,2,3 ; m = 0,1,2,3). Dans ce cas une orbitale ns, k orbitales np et m orbitales nd ont été « sommées ». Par rapport à chaque état d’hybridation, il y a une géométrie correspondante qui est la géométrie la plus stable. Cette dernière correspond à la distribution spatiale la plus stable des orbitales hybrides.
Remarque : pour faire des orbitales hybrides dans un même niveau d’énergie on commence d’abord par les orbitales ns et np avant les orbitales nd.
Les figures suivantes montrent des coupes du nuage électronique (diagramme de densité électronique) où plus les points sont denses, plus la probabilité de trouver cet électron est forte.
L'orbitale s est sphérique (l = 0).
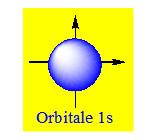
L'orbitale p (l = 1) est formée de 2 lobes centrés sur un axe commun. Il y a 3 orbitales p (m = -1, 0, +1), px suivant l'axe des x, py suivant l'axe des y et pz suivant l'axe des z.
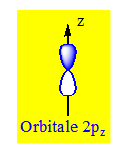
La forme des orbitales d (l = 2) est plus compliquée et il y a 5 orbitales d (m = -2, -1, 0, 1, 2) :
Trois orbitales d comportent 4 lobes qui se développent dans les plans bissecteurs des quadrants : (dxy, dxz, dyz).
Les deux autres orbitales d sont centrées sur les axes :
- dx2-y2 suivant les axes x et y,
- dz2 a 2 lobes centrés sur l'axe z et possède un petit volume torique dans le plan xOy.
Nous avons étudié précédemment les types de liaisons qui unissent des atomes au sein d'une molécule. A la lumière du modèle ondulatoire de la matière, l'on a pu montrer que chaque atome possède des orbitales atomiques. Les molécules possèdent également des orbitales, appelées orbitales moléculaires.
1-1 Origine
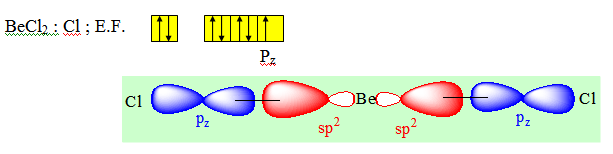
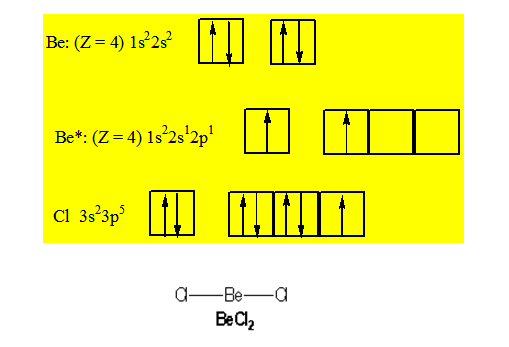
Considérons une molécule formée de trois atomes : BeCl2. Pour connaître les orbitales atomiques concernées dans la formation des deux liaisons entre l'atome de béryllium et les deux atomes de chlore, écrivons les structures électroniques de ces deux atomes.
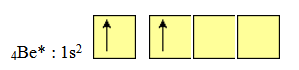
4Be : 1s22s2
Les deux liaisons portées par l'atome de Be ne peuvent être envisagées que pour une structure dite "excitée" de cet atome soit :
4Be* : 1s22s12p1 ; et en matérialisant la couche de valence avec les cases quantiques :
L’atome de chlore a la structure électronique: 17Cl : 1s22s22p63s23p5
Pour la couche de valence :
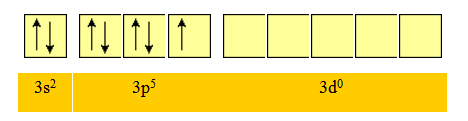
On peut former une première liaison covalente s entre l'orbitale atomique 2p de l’atome du béryllium et l'orbitale 3p du premier atome de chlore. Une telle liaison conduit à la formation d'une orbitale moléculaire localisée entre ces deux atomes ainsi qu'à un fort recouvrement.
Pour former la deuxième liaison Be — Cl nous disposons à nouveau d'une orbitale atomique 3p fournie par le deuxième atome de chlore. L'atome de béryllium ne dispose plus que d'une orbitale 2s de symétrie sphérique. La liaison construite à partir de ces orbitales atomiques s et p ne peut pas avoir de direction privilégiée. Cette liaison a aussi une plus faible énergie que la liaison précédente car le recouvrement 2s, 3p est plus faible que le recouvrement 2p, 3p. Or, ces résultats sont en désaccord avec l'expérience qui dit que les deux liaisons Be — Cl sont de même nature et caractérisées par la même énergie, la molécule BeCl2 est une molécule linéaire. En conclusion ce résultat montre que la construction des orbitales moléculaires de liaison à partir des orbitales atomiques des atomes ne peut être utilisée que pour les molécules diatomiques. Pour la molécule BeCl2 (3 atomes) on ne retrouve pas les données expérimentales (énergie de liaison - géométrie de la molécule). Si l'on veut rendre compte le mieux possible de la réalité il est nécessaire d'introduire une opération supplémentaire appelée l'hybridation des orbitales.
1-2 Principe de l’hybridation
Les orbitales atomiques sont des fonctions mathématiques. On sait que ces fonctions sont les solutions de l’équation différentielle (équation de Schrödinger) qui décrit le mouvement des électrons. Ces fonctions forment un espace vectoriel de fonctions car elles possèdent les propriétés caractéristiques des vecteurs d'un espace vectoriel : additivité, multiplication par un scalaire. Il est donc possible de construire à partir des deux orbitales atomiques 2s et 2p de l'atome de béryllium deux nouvelles orbitales h1 et h2 à partir des combinaisons linéaires :
h1 = a (2s) + b (2p)
h2 = a (2s) - b (2p)
Ces nouvelles orbitales atomiques sont appelées des orbitales hybrides. Les termes a et b sont des coefficients (scalaires) arbitraires.
1-2-1 Liaison simple et liaisons multiples
Il existe des liaisons simples, appelées liaison σ, et nous avons brièvement évoqué l'existence de liaisons multiples, notamment dans le cas du carbone, ces liaisons supplémentaires à σ, sont des liaisons π. Les liaisons σ sont des liaisons plus fortes que les liaisons π.
1-2-2 Le cas de la molécule d'hydrogène (H2)
La configuration de l'atome d'hydrogène est la plus simple : 1s1, l'orbitale d'un atome d'hydrogène est sphérique (s). Prenons deux atomes d'hydrogène :
Sachant que les orbitales sont des fonctions d'ondes, lors de la liaison, en assemblant ces deux volumes, on obtient l'orbitale moléculaire de H2.
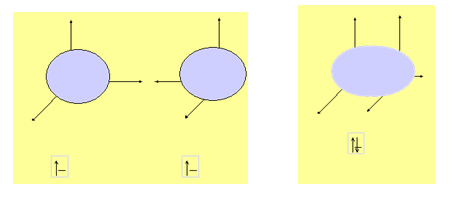
La molécule d'hydrogène a un nuage électronique composé de l'addition des deux volumes.
1-2-3 Liaison σ (p + s) : la molécule d'HCl

Définissons, par convention, l'axe sur lequel se fait la liaison comme étant l'axe Z :
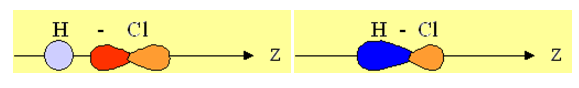
Il s'agit bien ici de liaison σ, celles-ci se font par recouvrement sur l'axe z. Passons aux liaisons π, c'est-à-dire lorsqu'il y a plus qu'une liaison entre deux atomes. Les liaisons π sont des liaisons moins fortes que les liaisons σ. Cela s'explique par le fait que le recouvrement des orbitales n'est pas tout à fait complet. Il s'agit pour les orbitales de liaisons π d'un recouvrement latéral, moins complet et moins efficace qu'un recouvrement sur l'axe correspondant :
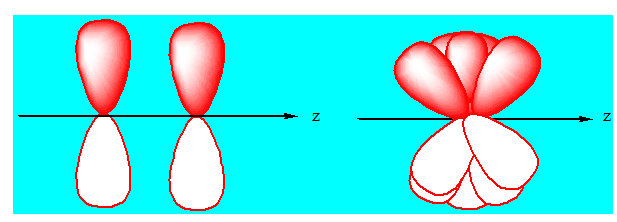
1-3 Hybridation sp
Les combinaisons linéaires étant de la forme :
h1 = a (2s) + b (2p)
h2 = a (2s) - b (2p)
Elles conduisent à deux nouvelles orbitales hybrides h1 et h2. Parmi l'infinité de combinaisons possibles, il existe une combinaison qui concentre la plus grande partie de l'hybride dans la direction de l'orbitale moléculaire que l'on veut construire. Dans le cas de BeCl2, plus l’orbitale hybride portée par le béryllium est concentrée dans la direction du chlore, plus le recouvrement avec l'orbitale atomique 3p de l'atome de chlore sera grand. Les combinaisons représentées ci-dessous sont conformes à cette condition.
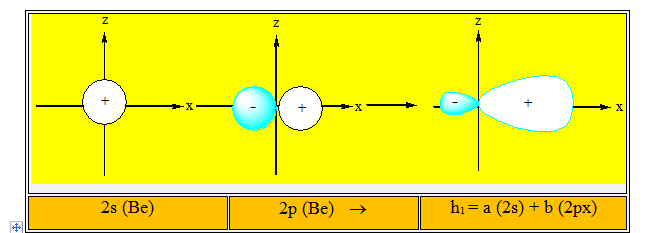
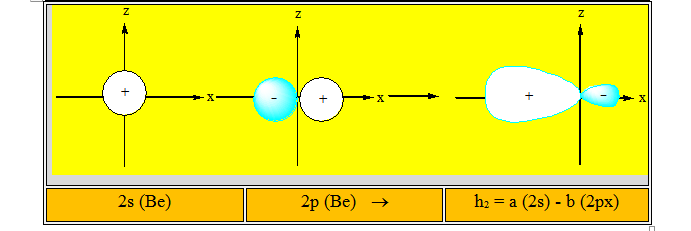
Les deux combinaisons linéaires conduisent à deux orbitales hybrides identiques mais dirigées dans deux directions opposées. Elles sont conformes à l'arrangement géométrique qui permet de construire avec chaque atome de chlore deux liaisons covalentes s identiques. Ces deux liaisons sont obtenues par le recouvrement de chaque orbitale atomique hybride de l'atome de béryllium avec une orbitale atomique 3p de chaque atome de chlore.
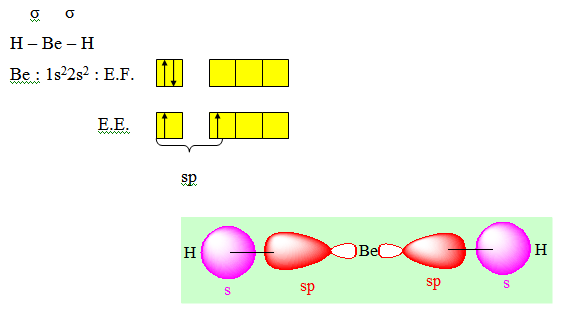
Ces deux liaisons ont la même énergie puisque les recouvrements sont les mêmes et elles forment entre elles un angle de 180°. La molécule obtenue est donc linéaire conformément à l'expérience. On dit que dans la molécule BeCl2 l'atome de Béryllium est dans l’état d’hybridation sp ou hybridé sp.
Autre exemple : BeH2 et BeCl2 à l’état de vapeur.
1-4 Hybridation sp2
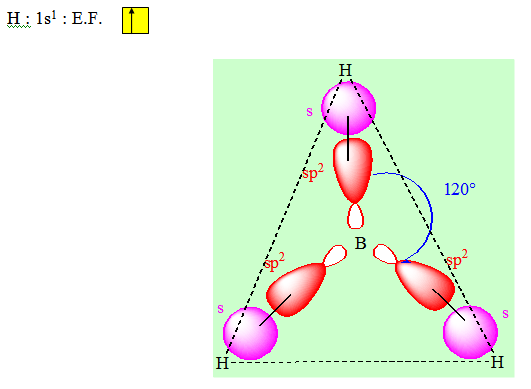
Considérons la molécule BF3 (trifluorure de bore). Cette molécule est plane, et les trois liaisons équivalentes BF forment entre elles des angles de 120° (règle de Gillespie).
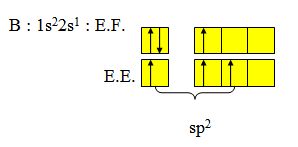
La structure électronique de l’atome de bore dans son état « excité » s'écrit :
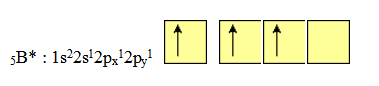
Pour construire les trois liaisons covalentes B — F, il faudra donc utiliser trois orbitales hybrides équivalentes, coplanaires et à 120°. Nous pouvons construire ces trois hybrides à partir de l'orbitale 2s et des orbitales 2px et 2py de l'atome de bore.
Exemple : BH3
Cependant il n’existe pas à l’état monomère ; il existe en dimère et sa formule est B2H6. Il y a 3 liaisons σ.
On a une distribution triangulaire.
H : 1s1 : E.F.
1-5 Hybridation sp3
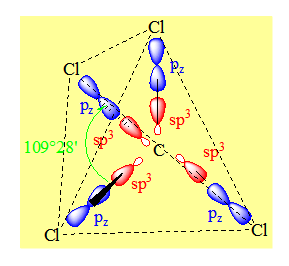
La molécule de méthane CH4 a une structure géométrique tétraédrique (angle du tétraèdre : 109°28'). Les quatre liaisons C-H de cette molécule sont équivalentes : longueur, énergie. Pour obtenir une telle structure il convient d'hybrider l'orbitale 2s et les trois orbitales 2p de l'atome de carbone.
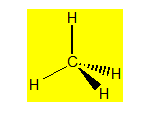
Chaque liaison C-H sera une liaison covalente s résultant du recouvrement entre une orbitale hybride sp3 de l’atome de carbone et l'orbitale 1s d’un atome d'hydrogène. Ces quatre liaisons sont équivalentes (même énergie) et dirigées selon les quatre sommets du tétraèdre.
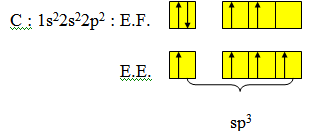
C’est une distribution tétraédrique.
1-6 Hybridation sp3d
1-6-1 Cas d’une bipyramide trigonale
Exemple du PF5. Il y a 5 liaisons σ autour de P. 
Les cinq orbitales hybrides seront distribuées en bipyramide trigonale. Un recouvrement de type axial entre les orbitales pz des fluors permet de rendre compte des liaisons σ.
Dans une bipyramide trigonale il y a 3 positions équatoriales équivalentes qui correspondent aux sommets du triangle. Les deux positions qui sont situées de part et d’autre du triangle sont appelées positions axiales ; elles sont aussi équivalentes. On a :
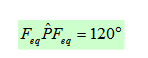
Si les atomes liants ne sont pas identiques alors ils sont répartis de manière à avoir le minimum de répulsion. Ainsi les atomes les plus gros ou les plus électronégatifs occupent toujours les positions équatoriales et ceux plus électropositifs les positions axiales.
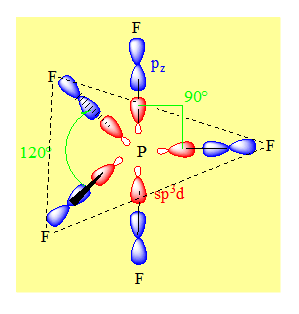
Remarque : Pour les éléments transitionnels la bipyramide trigonale est en général la géométrie la plus stable sauf pour l’antimoine Sb.
1-6-2 Cas d’une pyramide à base carrée
L’antimoine Sb et le phosphore ont le même nombre d’électrons sur la couche périphérique. On aura le même type de Lewis pour SbF5 et PF5. On aura aussi le même état d’hybridation pour l’élément central c’est – à – dire sp3d. Mais dans ce cas ci les orbitales hybrides sont distribuées suivant une pyramide à base carrée c’est la distribution spatiale la plus stable des orbitales hybrides. On fera un recouvrement de type axial entre les orbitales hybrides et les pz des fluors. Les angles valent 90°.
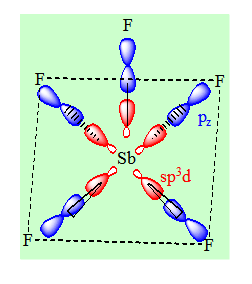
Remarques :
Si on a un anion dans lequel l’élément central est lié à 5 atomes et si la somme des électrons de l’élément central et d’un nombre d’électrons égal au nombre de charges de l’anion est égal au nombre d’électrons d’antimoine alors la géométrie de cet anion sera pyramidale à base carrée.
Il n’est pas possible sur la base de la théorie de l’hybridation de choisir entre une géométrie bipyramidale trigonale et une géométrie pyramidale à base carrée. Cependant pour les éléments non transitionnels la bipyramide trigonale est la géométrie la plus stable.
1-1 Hybridation sp3d2
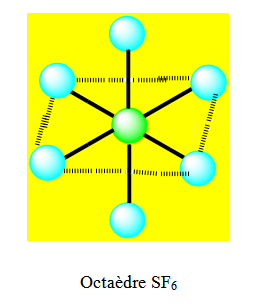
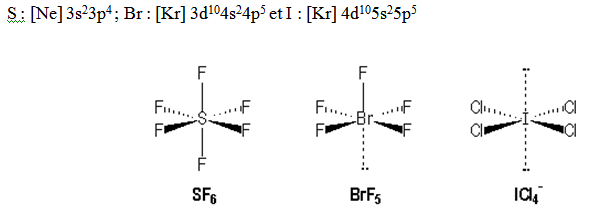
Exemple : SF6. Il y a 6 liaisons σ autour de S.
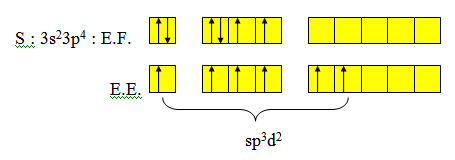
Six liaisons σ d’où l’hybridation est sp3d2. La distribution spatiale la plus stable est la distribution octaédrique. Tous les angles valent 90°. On ferait un recouvrement de type axial avec pz pour les liaisons σ. Les atomes les plus gros et les plus électronégatifs sont en position trans.
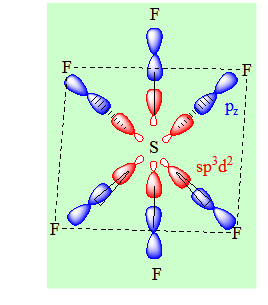
1-8 Hybridation sp3d3
Exemple : IF7. Il y a 7 liaisons σ autour de I.
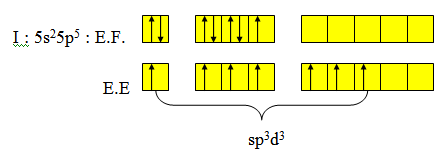
Sept liaisons σ ; l’hybridation sera sp3d3. La distribution spatiale la plus stable est la bipyramide pentagonale. Pour les liaisons σ on aura un recouvrement de type axial entre les orbitales hybrides et les pz des fluors. Contrairement à une bipyramide trigonale dans le cas d’une bipyramide pentagonale les atomes liants les plus gros ou les plus électronégatifs sont placés en position axiale.
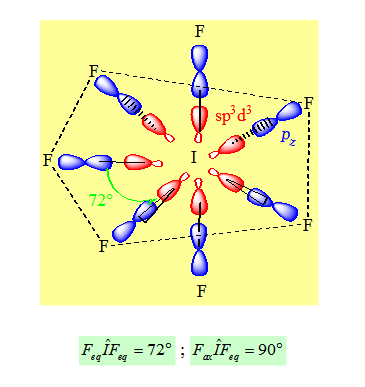
Remarque :
Dans une hybridation sp3d, sp3d2 ou sp3d3 les orbitales n dx2-y2 et n dz2 sont prioritaires.
Contrairement à une bipyramide, dans le cas d’une bipyramide pentagonale les atomes liants les plus gros ou les plus électropositifs sont placés en position axiale.
1-9 Types d'hybridation et liaisons π
1-9-1 Hybridation sp2 avec liaison π
Une liaison π est obtenue par recouvrement de deux orbitales parallèles. On peut avoir deux orbitales parallèles p ou une orbitale p et une orbitale d.
Exemple : l’éthylène.
On a 3 orbitales hydrides pour chaque carbone.
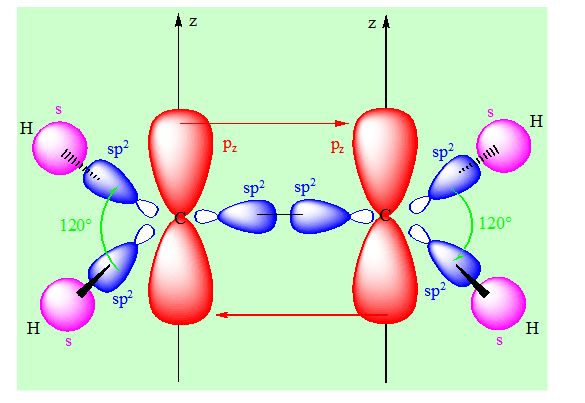
L’hybridation des carbones est sp2. Un recouvrement de type axial entre deux orbitales hybrides de chaque carbone avec les orbitales s des hydrogènes d’une part ; entre les deux orbitales restantes d’autre part permet de rendre compte du squelette σ de l’éthylène c’est – à – dire de l’ensemble des liaisons présentes dans cette molécule. Après l’hybridation sp2 ; il reste sur chaque carbone une orbitale pz. Les deux orbitales pz sont parallèles et donnent un recouvrement de type latéral avec une liaison π. Les angles sont de 120°. La liaison π entraîne que la molécule est plane globalement.
1-9-2 Hybridation sp avec liaison π
Exemple : l’acétylène

On a deux liaisons σ.
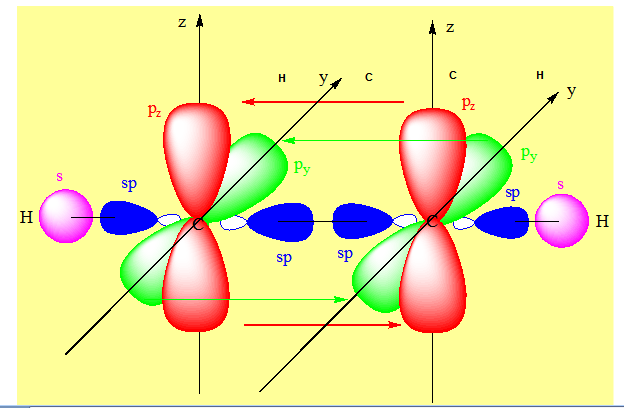
py et pz de l’un des carbones sont respectivement parallèles aux orbitales de même nom de l’autre carbone et donnent par recouvrement de type latéral les liaisons π. Exemple du dioxyde de carbone.
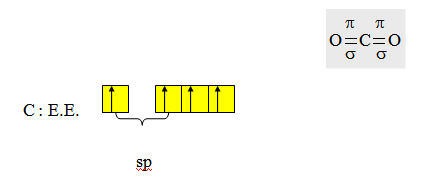
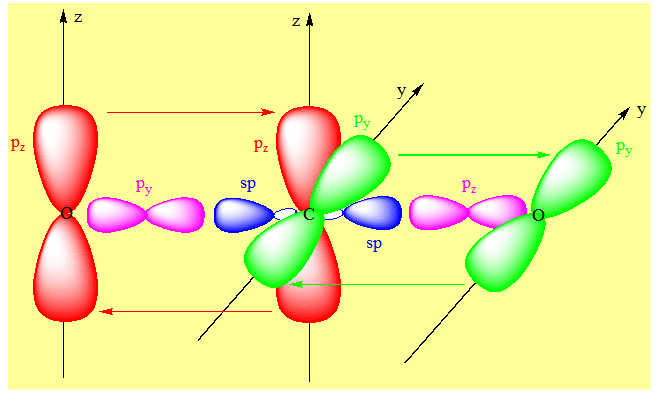
L’état d’hybridation du carbone est sp ; on peut utiliser les orbitales py ou pz de l’oxygène. Après l’hybridation du carbone il reste une orbitale py et une orbitale pz ; il faudrait donc que les orbitales qui restent sur les deux oxygènes après qu’on ait rendu compte du squelette σ soient une orbitale py et une orbitale pz de façon à ce qu’elles respectivement parallèles à py et pz du carbone. On se rend compte en prenant des orbitales de même nom pour les liaisons π sur les oxygènes ; elles ne pourront pas être parallèles à des orbitales de noms différents sur le carbone. Par recouvrement de type latéral ces orbitales donneront des liaisons π.
1-9-3 Hybridation sp3 avec liaison pπdπ : Phénomène de résonance
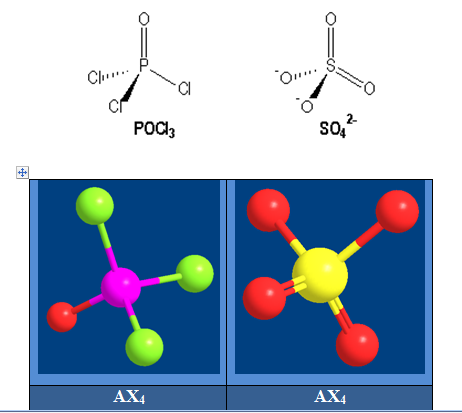
Exemple : l’ion sulfate
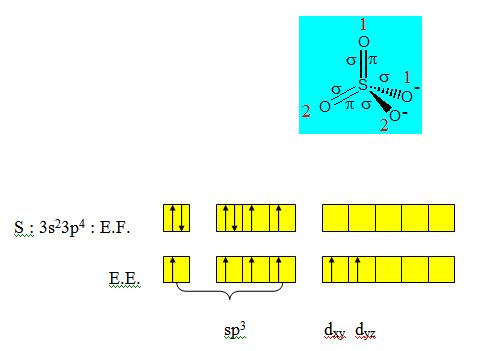
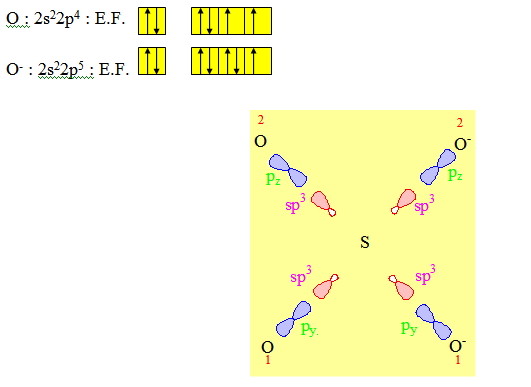
 Après l’hybridation de sp3 il restera 2 électrons sur les orbitales d. Ce sont ces orbitales qui seront utilisées pour les liaisons π comme dans le cas de C2H4 ; C2H2 et CO2.
Après l’hybridation de sp3 il restera 2 électrons sur les orbitales d. Ce sont ces orbitales qui seront utilisées pour les liaisons π comme dans le cas de C2H4 ; C2H2 et CO2.
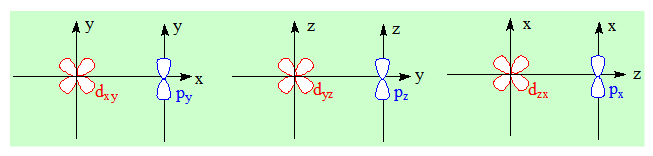
Rappelons que seules 3 des 5 orbitales d peuvent donner des liaisons π ; elles sont appelées orbitales mixtes : dxy ; dyz et dzx.
dxy est parallèle à py; dyz est parallèle à pz et dzx est parallèle à px.
Pour le recouvrement de type axial ; nous utiliserons pour les oxygènes non chargés 2 orbitales de noms différents (py et pz) de sorte que les orbitales restantes (pz et py) soient respectivement parallèles à dyz et dxy qui sont nécessairement dans ce cas les 2 orbitales d qui reçoivent les électrons venant de la double promotion électronique. Un recouvrement de type latéral entre ces orbitales d d’une part et p de l’autre permet de rendre compte des liaisons π dans SO42-. Ces liaisons sont ainsi appelées liaisons pπdπ ; le nom venant du fait que l’une des orbitales de la liaison π est une orbitale d et l’autre une orbitale p.
Les 4 oxygènes du sulfate sont normalement tous équivalents. On peut les numéroter 1 ; 2 ; 3 ; 4 et leur faire porter à tour de rôle la charge et la double liaison ; nous aurons 4 structures résonantes limites qui sont :
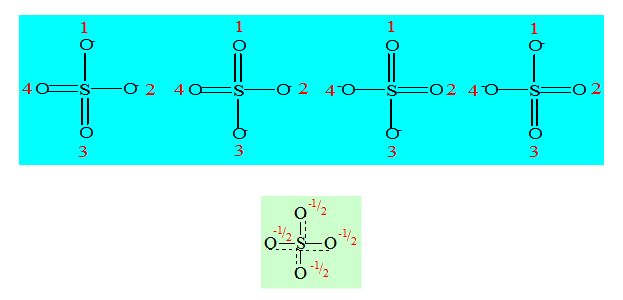
Est la structure vraie qui sera une structure moyenne qui tiendra compte de ces 4 structures limites. Tout se passe comme si les liaisons π étaient partagées entre les 4 oxygènes et les 2 charges aussi. L’ordre de liaison serait donc 1,5 pour chaque S-O puisqu’à côté de la liaison σ il y a une moitié de liaison π et une charge – ½ sur chaque oxygène ce qui confère à l’ion sulfate une structure rigoureusement tétraédrique. On dit que les oxygènes sont équivalents par phénomène de résonance ; ceci est applicable à tous les oxoanions. On a toujours ce phénomène dans un oxoanion. L’ordre de liaison entre l’atome central et les oxygènes sera donné par :

Conclusion : La théorie de l’hybridation est importante car elle nous permet de comprendre la structure des molécules simples dont l’élément central n’a pas de doublets libres. C’est un procédé mathématique qui permet de mieux expliquer une structure donnée. Cependant cette théorie a des limites car elle ne permet pas d’une part de trancher entre une bipyramide trigonale et une pyramide à base carrée pour les molécules AX5 et d’autre part ne permet pas de prévoir la géométrie des molécules dont l’élément central possède un ou plusieurs doublets. Cependant ces limites seront dépassées, du moins en partie, par la théorie des orbitales moléculaires.
2 Le modèle de la répulsion des charges ou modèle de Gillespie (VSEPR)
Ce modèle, très simple, ne nécessite pas l’utilisation de la théorie quantique ; il ne fait donc pas intervenir les constructions d’orbitales atomiques. Il se contente, comme base de départ, la notation de Lewis pour les atomes. L’élément central possède des paires libres ou pas.
2-1 Origine
En 1957 le chimiste canadien R.J Gillespie (université Mc Master Hamilton, Ontario) reprenant une idée émise par les britanniques N. Sigdwick et H. Powell, a développé les règles de la théorie de répulsion des électrons de la couche de valence (VSEPR de Valence Shell Electron Pair Repulsion). La méthode VSEPR peut être considérée comme le prolongement dans le domaine stéréochimique de la description de la liaison chimique par appariement électronique de G.N Lewis (1916). Elle permet de prévoir la géométrie de toutes les molécules simples.
Les électrons de la couche de valence de la plupart des molécules et des ions stables sont en nombre pair (il existe des molécules possédant un nombre impair d'électrons comme NO2 mais cela reste assez rare). Le principe de Pauli (1920) nous indique que seuls les électrons possédant des nombres quantiques de spin opposés évoluent dans une même région de l'espace car ils sont décrits par la même orbitale. Ces électrons peuvent être groupés par paires ou doublets.
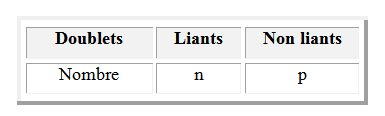
Dans le modèle de Lewis, la liaison entre deux atomes est décrite par la mise en commun d'un doublet d'électrons : on l'appelle doublet liant. Les paires d'électrons non utilisées pour décrire les liaisons constituent les doublets non liants notés E.
2-2 Principes de base
L'approximation de départ de la théorie consiste à assimiler les paires d'électrons à des charges électriques ponctuelles. Celles de la couche de valence évoluent à une distance comparable du noyau de l'atome. En première approximation, elles sont assujetties à se déplacer sur une sphère dont le centre est occupé par le noyau. L'arrangement géométrique qu'elles adoptent sur cette sphère est la conséquence de leur répulsion mutuelle.
Bien qu'il ne s'agisse pas à proprement parler d'une interaction électrostatique entre charges ponctuelles, car le comportement des électrons soit régi par les lois de la mécanique quantique, on obtient un résultat correct en recherchant l'arrangement qui rend maximum les distances entre les paires d'électrons.
A ce premier niveau d'approximation, il n'y a pas de distinction à faire entre un doublet liant et un doublet non liant.
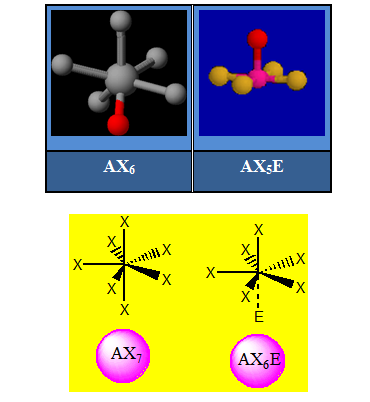
Dans ces conditions, l'arrangement qui minimise la répulsion des doublets dépend seulement de leur nombre : n + p. Cela correspond aux figures géométriques suivantes en se limitant à la coordination sept. Si p = 0 l’environnement est égal à la géométrie ; si p ≠ 0 l’arrangement est différent de la géométrie.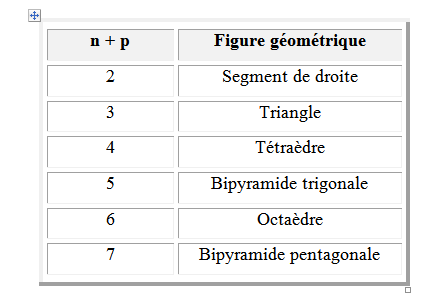
A ces six types de figures géométriques, correspondent davantage d'arrangements des paires d'électrons que nous allons passer en revue. Dans ce qui suit X désigne un atome et E un doublet non liant.
En première approximation, les liaisons multiples sont traitées comme si elles ne faisaient intervenir qu'un seul doublet.
Nous prendrons des exemples de molécules avec des liaisons simples et des molécules avec des liaisons multiples.
Valence Shell Electron Pair Repulsion Theory (ou théorie de la répulsion des paires d’électrons dans la couche de valence).
Principe : les paires électroniques liantes et libres se repoussent mutuellement. Par conséquent, elles se localisent dans l'espace de manière à minimiser leur répulsion.
Ce modèle permet d'expliquer la géométrie des paires électroniques autour d'un atome central. A une même géométrie, plusieurs formes de molécules pourront correspondre.
2-3 Règles et notations VSEPR
Le composé a pour formule AXnEp ; où A est l'atome central de la molécule, X représente les paires liantes, E les paires libres et n, p représentent le nombre de paires de chaque type. Pour déterminer la géométrie d’une molécule par la théorie VSEPR :
On commence par déterminer la structure de Lewis de la molécule ;
On compte le nombre de paires libres et de paires liantes ;
On écrit la molécule sous la forme AXnEp ;
Le nombre total de paires n + p donne la distribution spatiale la plus stable. Cet arrangement indique l’environnement de l’élément central.
Règle n°1 : Il faut minimiser la répulsion entre les paires électroniques (libres et liantes). Celles-ci se répartissent l'espace disponible. Donc les n paires d’électrons se répartissent dans l’espace de façon à assurer une répartition maximale.
Règle n°2 : Les paires libres occupent plus de place que les paires liantes car la répulsion des paires libres est plus forte. Les angles entre les paires seront différents. De plus si les différents endroits ne sont pas équidistants, les paires libres se placeront préférentiellement le plus loin.
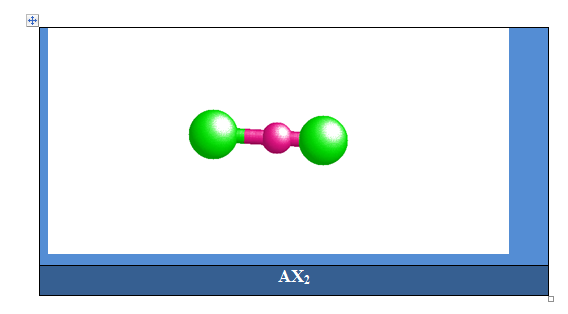
Cas des molécules avec 2 paires électroniques
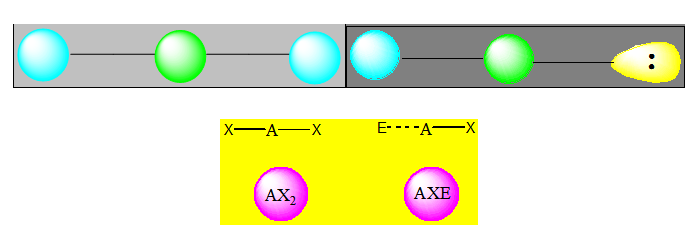
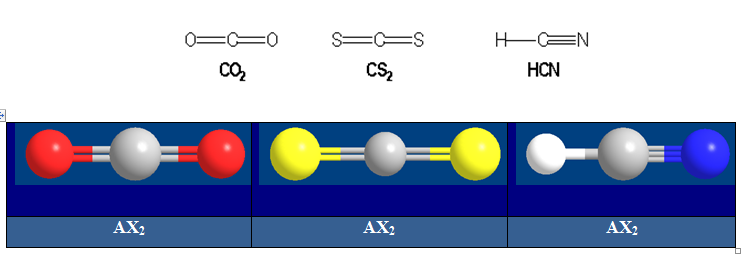
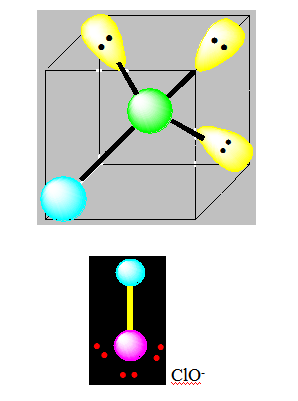
Deux paires opposées au maximum (180°). La molécule est linéaire et l'angle entre les deux paires est un angle plat (180°). L’arrangement et la géométrie de ces types de molécules sont linéaires. Ce sont les molécules de type AX2. Le béryllium, élément de la seconde colonne de la classification périodique de configuration électronique 1s22s2, est lié par 2 liaisons simples à deux atomes de chlore dans la molécule BeCl2.
Molécules avec des liaisons multiples :
Cas des molécules avec 3 paires électroniques
On distingue trois familles. Les molécules peuvent être de type AX3 ou de type AX2E ou AXE2.
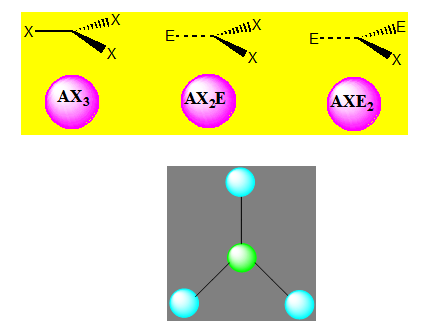
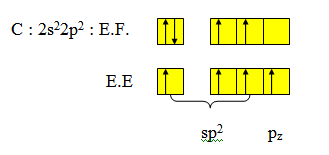
Trois paires situées dans le même plan et à égale distance les unes des autres : 120° (sp2), La molécule a la forme d'un triangle équilatéral. La molécule est triangulaire (exemple BF3).
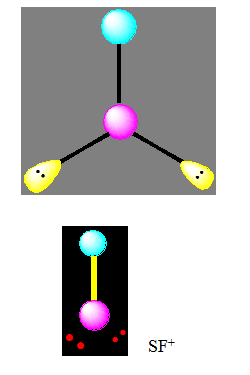
La molécule de type AX2E est angulaire mais l’arrangement reste triangulaire. L’angle est voisin de 120° (exemple SnCl2).
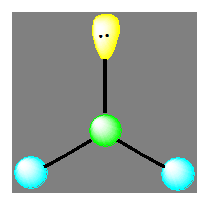
Molécules avec des liaisons simples :
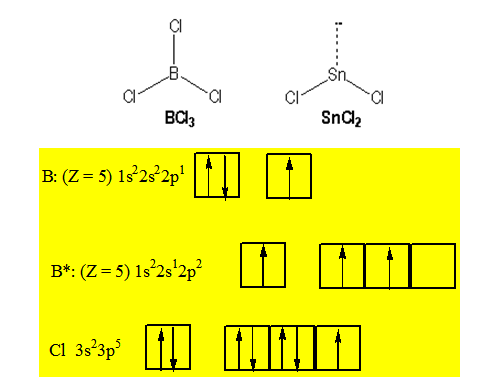
B : [He] 2s22p1
Sn : [Kr] 4d105s25p2
La diminution de l’angle de 120° à 109° est due à la présence de la paire libre qui a tendance à occuper beaucoup plus d’espace.
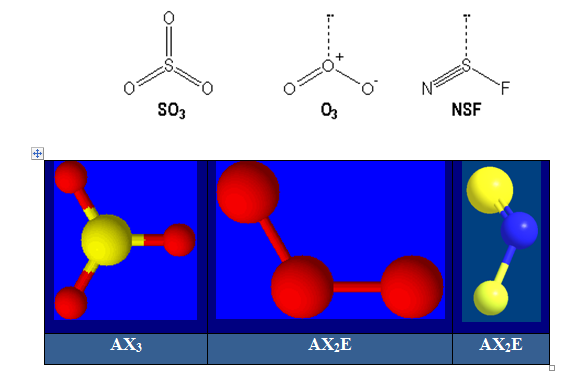
Molécules avec des liaisons multiples :
S : [Ne] 3s23p4 ; O : [He] 2s22p4 et N : [He] 2s22p3
Cas des molécules avec 4 paires électroniques
On distingue quatre familles : AX4 ; AX3E ; AX2E2 et AXE3.
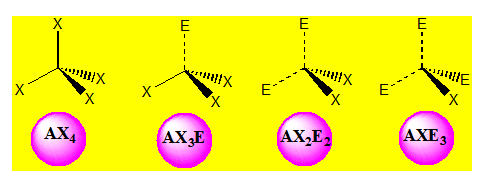
Pour AX4 on a : 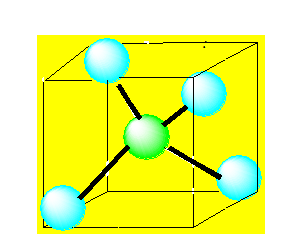
La molécule est tétraédrique. Quatre paires liantes dirigées vers le sommet d'un tétraèdre régulier et située à 109° les unes des autres. La molécule est de forme tétraédrique. Donnons l’exemple du méthane CH4.
Pour AX3E on a une distribution tétraédrique l’angle vaut 107° avec l’ammoniac NH3 ; la géométrie est pyramidale.
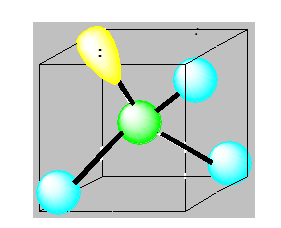
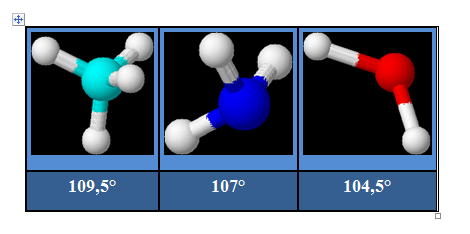
Pour AX2E2 la distribution est tétraédrique la géométrie est en « V ». ou angulaire ou coudée. Pour H2O l’angle vaut 104,5°.
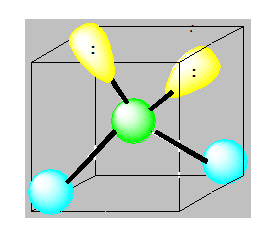
Pour AXE3 l’arrangement est tétraédrique et la géométrie est linéaire.
Molécules avec des liaisons simples :
Si : [Ne] 3s23p2 ; P : [Ne] 3s23p3 et O : [He] 2s22p4
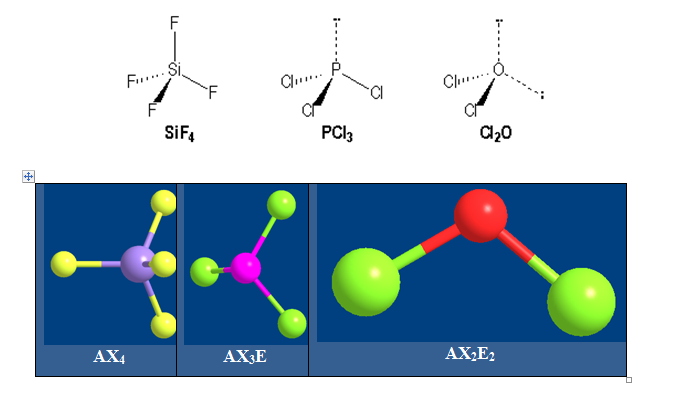
Les angles diminuent quand on passe de AX4 à AX2E2. Car le nombre de paire libres augmente autour de l’élément central.
Molécules avec des liaisons multiples :
P : [Ne] 3s23p1 et S : [Ne] 3s23p4
Cas des molécules avec 5 paires électroniques
La figure géométrique la plus répandue et qui est la plus stable correspondant à cet arrangement est la bipyramide trigonale. Une complication apparaît ici du fait de la non équivalence des positions aux sommets de la bipyramide. On peut les classer en deux catégories : axiales (a) et équatoriales (e) :
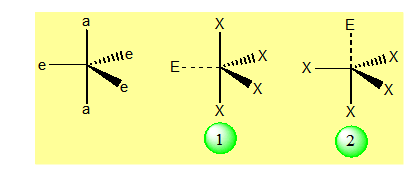
Le problème peut être résolu en affinant les hypothèses de la méthode. A un deuxième niveau d'approximation les paires liantes et non- liantes sont traitées différemment : la position dans l'espace des électrons constituant un doublet liant est contrôlée par le champ exercé par les noyaux de deux atomes, celle des électrons d'un doublet non liant par un seul noyau, celui de l'atome central. Il est donc raisonnable d'admettre que le volume moyen dans lequel évoluent les électrons d'un doublet non liant, est supérieur à celui dont disposent les électrons d'un doublet liant. Ces considérations simples permettent d'établir la hiérarchie suivante dans la répulsion entre les paires :
![]()
Un paramètre pratique pour évaluer l'ordre de grandeur de la répulsion entre les doublets, est l'angle au centre a formé par les directions entre ceux-ci et l'atome central. En utilisant ce paramètre, on peut comparer les énergies des deux arrangements (1) et (2) en décomptant les interactions sur la base des inégalités précédentes en tenant compte seulement des interactions pour lesquelles a £ 90° les autres étant supposées négligeables.
Pour l’arrangement (1) le doublet E interagit avec 2 atomes X ;
Pour l’arrangement (2) le doublet E interagit avec 3 atomes X.
Le premier arrangement est donc plus stable que le second. A cet arrangement correspond une catégorie de molécules qu'on peut appeler avec Gillespie molécules tétraédriques déformés et à laquelle appartient SF4. Expérimentalement, on ne connaît pas de stéréo-isomère de cette molécule.
Une étude du même type effectuée pour les autres arrangements a priori possibles en géométrie pentagonale, montre que les paires électroniques non liantes et les liaisons multiples (double ou triple) et les gros substituants doivent occuper de façon préférentielle les positions équatoriales où elles occupent le plus d’espace.
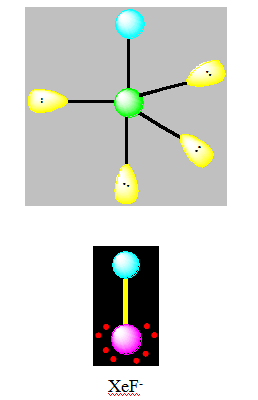
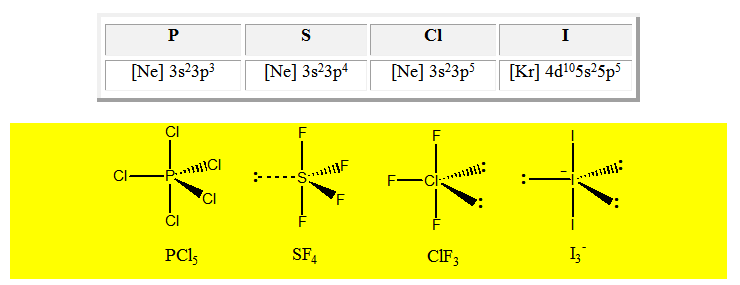
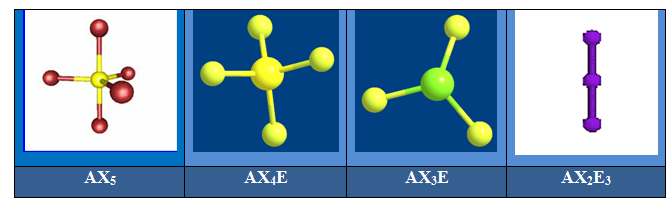
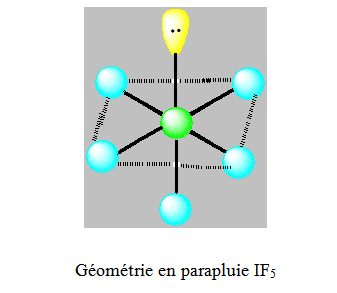
On distingue cinq familles : AX5 ; AX4E ; AX3E2 ; AX2E3 et AXE4. Dans une bipyramide trigonale les paires libres sont toujours placées en position équatoriale où elles occupent le plus d’espace.
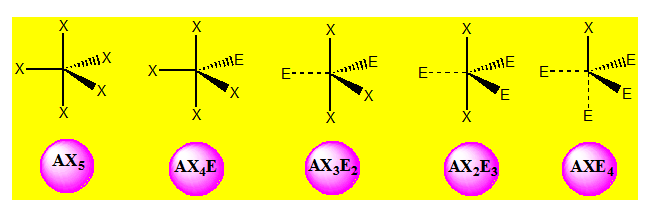
Cinq paires. Les paires forment entre elles un angle de 120°. La géométrie de PCl5
est bipyramidale trigonale.
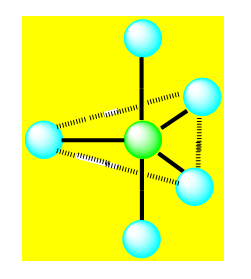
Pour AX4E la géométrie est un tétraèdre déformé. Exemple SF4.
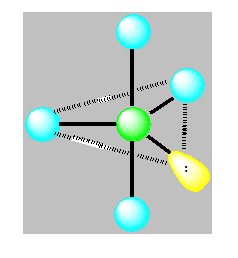
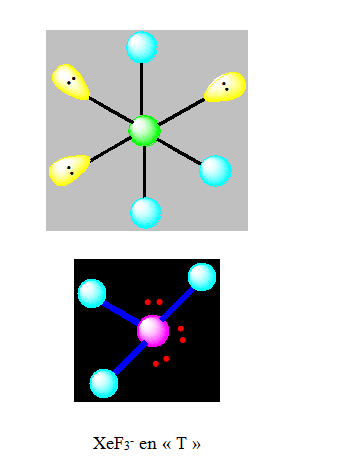
Pour AX3E2 la géométrie est en pic ou en forme de « T ». Exemple ClF3.

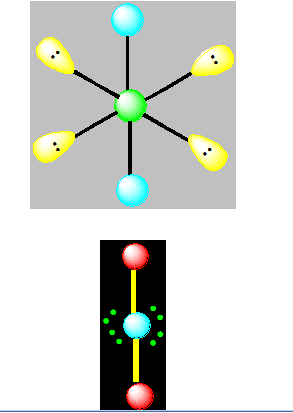
Pour AX2E3 la géométrie est linéaire. C’est l’exemple de XeF2.
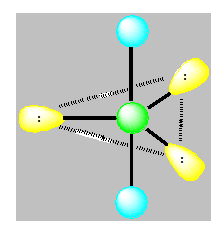
Pour AXE4 la géométrie est linéaire.
Molécules avec des liaisons simples :
Molécules et ions avec des liaisons multiples :
S : [Ne] 3s23p4 et I : [Kr] 4d105s25p5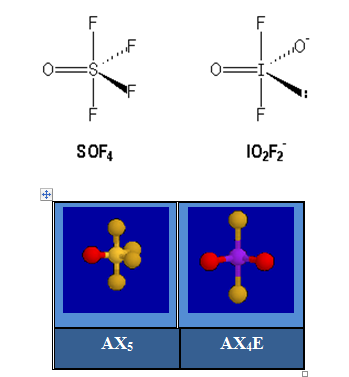
NB : L’antimoine Sb a tendance à avoir un environnement pyramidal à base carrée (exemple SbF5).
Cas des molécules avec 6 paires électroniques
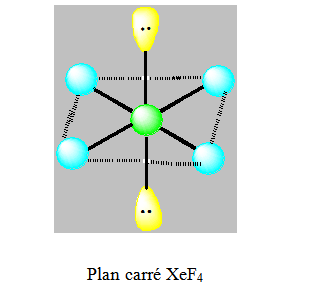
On distingue les cinq familles suivantes : AX6 ; AX5E ; AX4E2 ; AX3E3 et AX2E4.
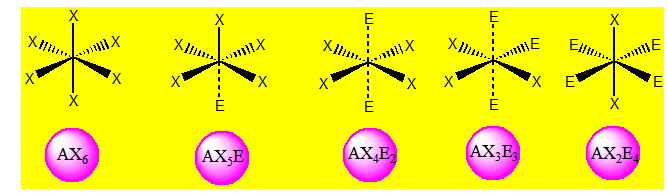
Six paires. Les paires du plan équatorial forment entre elle un angle de 90°. Les paires axiales formant un angle de 120° avec les autres. Les paires libres qui occupent dans l’octaèdre des positions différentes mais géométriquement équivalentes doivent se placer le plus loin possible l’une de l’autre. Ainsi on dit que ces paires libres sont en position trans et les paires liantes en position cis.
Molécules avec des liaisons simples :
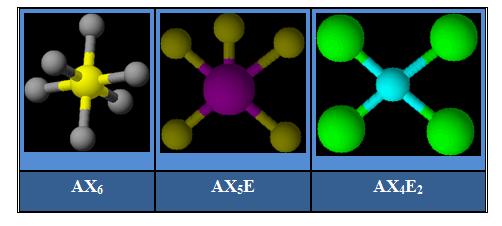
Molécules avec des liaisons multiples : Dans une molécule quand on a une liaison multiple ; on la considère comme étant une paire libre parce qu’une paire libre est plus grosse qu’une paire liante.
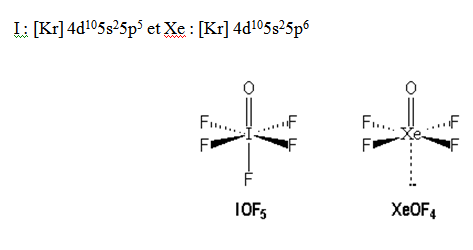
2-4 Cas des molécules avec liaisons multiples
Pour les bipyramides trigonales les liaisons multiples et les paires libres se mettent toujours en position équatoriale pour minimiser les répulsions car elles occupent plus d’espace. De même les atomes les plus gros et les plus électropositifs occupent les positions équatoriales.
Pour les pyramides à base carrée les liaisons multiples et les paires libres se mettent toujours en position axiale. De même les atomes plus électronégatifs occupent ces positions.
2-5 Cas des molécules ioniques
Dans le cas des ions on suit les mêmes règles que dans celui des molécules neutres.
Pour déterminer la structure des anions les charges négatives sont affectées aux atomes les plus électronégatifs sauf dans le cas des oxoanions où les charges sont portées par les atomes d’oxygène.
Dans le cas des cations les charges positives sont portées par les atomes les plus électropositifs.
2-6 Effets du second ordre
Influence des paires libres sur les angles standards de liaisons
A un deuxième niveau d'approximation, il est logique d'admettre que les doublets non liants, moins confinés dans l'espace internucléaire que les doublets liants, occupent un volume moyen plus élevé. On interprète ainsi la diminution progressive de l'angle entre les liaisons quand on passe de CH4 à NH3 puis H2O. La paire libre occupant plus de place ; les paires liantes vont se rapprocher pour lui faire de la place ce qui explique la fermeture de l’angle solide de l’azote (107°) au lieu de 109° dans le tétraèdre régulier.
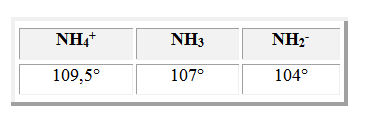
On observe une évolution du même type pour les dérivés ioniques de l'ammoniac :
Arrangement triangulaire :
Dans les molécules de HCHO et de COCl2 représentées ci-dessous, l'angle entre les liaisons simples est inférieur à 120°.
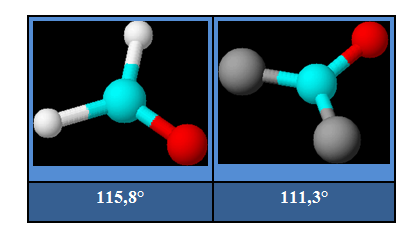
Arrangement tétraédrique :
On assiste à la fermeture de l'angle entre les liaisons simples au fur et à mesure que l'ordre de liaisons croît dans la série : SiF4, POF3, NSF3.
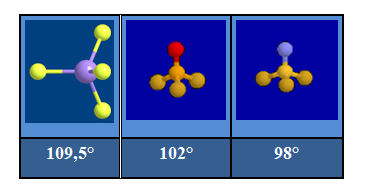
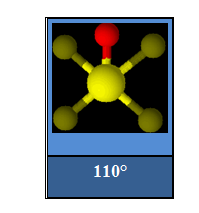
Arrangement pentagonal :
L'angle FSF est inférieur à 120° dans SOF4.
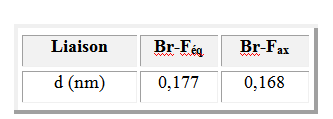
Si nous prenons le cas de ClF3 ; nous avons une bipyramide trigonale avec 2 paires en position équatoriale ; l’angle FaxClFeq = 87° au lieu de 90° à cause des 2 paires libres.
Arrangement octaédrique :
Si on prend le cas de BrF5 ; l’angle FaxBrFeq = 85° au lieu de 90°.
Influence du volume des liaisons multiples
La forme géométrique globale ne dépend que des liaisons s. On peut donc classer les molécules possédant des liaisons multiples dans les mêmes groupes que celles qui possèdent des liaisons simples. En revanche le volume occupé par les électrons dans l'espace croit avec l'ordre de liaison. On assiste donc à une fermeture de l'angle opposé à la liaison multiple.
Influence de l’électronégativité
Le volume occupé par les doublets liants décroît lorsque la différence d'électronégativité des atomes liés augmente. Drainés vers l'atome le plus électronégatif, les électrons de liaison davantage confinés dans l'espace internucléaire se repoussent moins. Pour un même atome central on observe une diminution de l'angle entre les liaisons avec l'accroissement de l'électronégativité de l'atome périphérique. L'évolution des angles dans la série : PI3, PBr3, PCl3 et PF3 en témoigne.
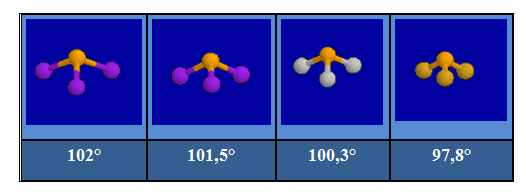
Les molécules NH3, PH3 et AsH3 ont chacune trois atomes d'hydrogène et des atomes centraux de moins en moins électronégatifs.
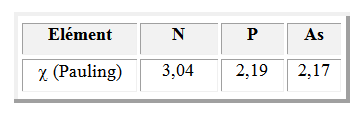
En passant de NH3 à PH3 puis AsH3, la mise en commun des électrons de liaison est de plus en plus importante et les doublets de liaisons occupent de moins en moins de place dans l'espace. On observe une diminution de l'angle au sommet de la pyramide.
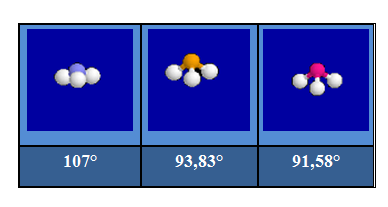
Combinaison des effets de volume et d'électronégativité
Les effets décrits précédemment peuvent naturellement s'ajouter ou se compenser partiellement. Dans les molécules C2H4 et C2F2H2, les angles opposés à la double liaison sont inférieurs à 120°. On l'interprète de la façon suivante : le volume occupé par les électrons de la liaison double est supérieur à celui occupé par les électrons d'une liaison simple. Dans C2F2H2 cet angle est inférieur à celui observé dans C2H4 car les liaisons C-F fortement polarisées, occupent moins de place que les liaisons C-H.
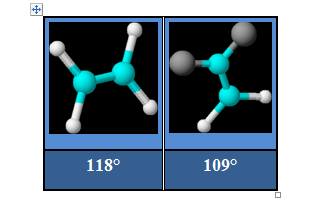
Non équivalence des positions axiales et équatoriales en géométrie pentagonale
On sait que les positions axiales et équatoriales ne sont pas équivalentes dans des molécules comme PF5 ou PCl5. L'interaction impliquant des doublets non liants en position équatoriale est moindre qu'en position axiale. Il en résulte une diminution de la longueur des liaisons équatoriales et un allongement des liaisons axiales.
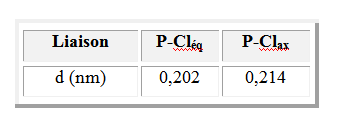
La non équivalence entre position axiale et équatoriale se manifeste également pour les doublets liants. Dans la molécule PF3Cl2, Cl est moins électronégatif que F. Le doublet assurant la liaison entre P et Cl occupe une place plus grande que celui entre P et F. On explique ainsi que l'isomère dans lequel ces atomes sont en position équatoriale soit le plus stable.
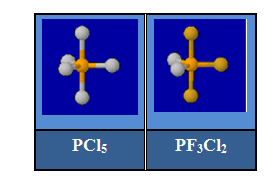
Pour les molécules de type AX5E comme BrF5, on observe une légère diminution de l'angle entre les liaisons (85° au lieu de 90°) et une liaison équatoriale plus longue que la liaison axiale.
2-7 Limites de la méthode VSEPR
La théorie VSEPR est une méthode qui permet de prévoir les géométries de toutes les molécules dont l’élément central est non transitionnel ou s’il est transitionnel sa couche interne est nd5 ou nd10. Cette méthode permet la plupart du temps la prédiction correcte de l'arrangement local des paires d'électrons autour d'un atome lorsque celui-ci peut être choisi sans ambiguïté comme atome central. En revanche, son application est beaucoup plus problématique quand il s'agit de prévoir la géométrie globale de molécules complexes. Ainsi il n'est pas possible de prévoir la planéité de la molécule d'éthylène ni d'expliquer pourquoi chez les allènes ; les substituants sont situés dans des plans perpendiculaires. Par ailleurs les molécules ne sont pas des objets statiques. Cela est particulièrement net dans des molécules faisant intervenir des atomes pyramidaux comme NH3, PH3. Il existe aussi des phénomènes plus complexes comme l'échange des positions axiale et équatoriale en géométrie pentagonale.
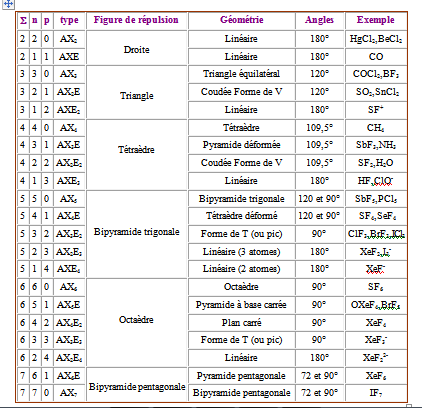
Tableau récapitulatif des différents arrangements des doublets de tous les cas rencontrés
3 Géométrie des complexes de coordination
Pour déterminer la géométrie des complexes on procède de la même façon que dans le cas de la théorie de l’hybridation :
- On cherche autant d’orbitales vides que de liaisons datives autour de l’élément centrale ;
- On somme les orbitales vides de l’élément central ;
- L’hybridation du métal central dépend de la nature des ligands qui l’entourent. La série spectroscopique de Fajans – Tsuchida classe les ligands en fonction de la force de leur champ.
CO > CN- > NO2- > éthylène diamine > NH3 > CH3CN >NCS- >H2O > C2O42- > OH- > F- > NO3- > Cl- > SCN- > S2- > Br- > I-. (En rouge les ligands à champ fort, en bleu les ligands à champ moyen et en noir ceux à champ faible).
- Si les ligands sont à champ fort (à bas spin) les électrons libres de l’orbitale (n – 1)d sont couplés. Dans ce cas on somme dans l’ordre (n – 1)d, ns, np les orbitales vides de l’élément central ; on obtient ainsi les hybridations suivantes : dsp2, dsp3, d2sp, d2sp2 ou d2sp3. On obtient le même phénomène si l’élément central appartient à la deuxième et à la troisième série des éléments de transition. Dans ce cas, on obtient des complexes diamagnétiques si le nombre d’électrons nd est pair.
- Si les ligands sont à champ faible (à haut spin), les électrons libres de l’orbitale (n – 1)d ne sont pas couplés. Dans ce cas on somme dans l’ordre ns, np, nd les orbitales vides de l’élément central ; on obtient ainsi les hybridations : sp3, sp3d, sp3d2. Dans ce cas, on obtient des complexes paramagnétiques.
Les géométries des complexes selon le type d’hybridation de l’élément central sont regroupées sur le tableau ci – dessous.
|
Complexes |
Nombre de liaisons |
Hybridation |
Géométrie |
|
AL4 |
4 |
sp3 |
Tétraèdre |
|
AL4 |
4 |
dsp2 |
Plan carré |
|
AL4 |
4 |
d2sp |
Plan carré |
|
AL5 |
5 |
sp3d |
Bipyramide trigonale |
|
AL5 |
5 |
dsp3 |
Pyramide à base carrée |
|
AL5 |
5 |
d2sp2 |
Pyramide à base carrée |
|
AL5 |
5 |
d3sp |
Pyramide à base carrée |
|
AL6 |
6 |
sp3d2 |
Octaèdre |
|
AL6 |
6 |
d2sp3 |
Octaèdre |
|
AL6 |
6 |
dsp3d |
Octaèdre |
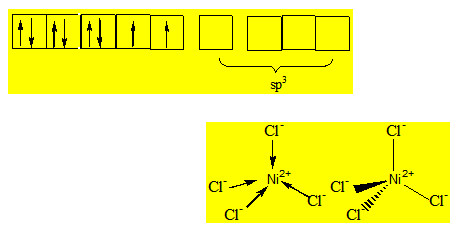
Exemple 1 : Géométrie de NiCl42- :
Ni2+ : (Z = 28) [Ar] 3d8. Les ions Cl- étant des ligands à champ faible on somme l’orbitale 4s et les 3 orbitales 4p ; Ni2+ est hybridé sp3 ; la géométrie du complexe est un tétraèdre.
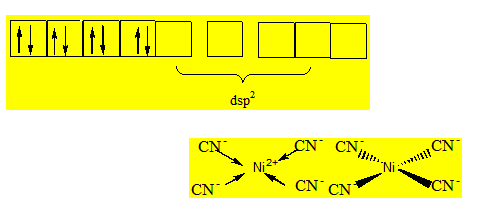
Exemple 2 : Géométrie de Ni(CN)4- :
Les ions CN- étant des ligands à champ fort tous les électrons de l’orbitale 3d sont couplés. On somme dans le métal central l’orbitale vide 3d ; l’orbitale 4s et deux orbitales 4p : Ni2+ est hybridé dsp2 ; la géométrie du complexe est un plan carré.
Exemple 3 : Géométrie de [Os(NH3)5]2+
Os2+ (Z = 76) : 1s22s22p63s23p64s23d104p65s24d105p64f145d6. Il est hybridé d2sp2 ; la géométrie du complexe est une pyramide à base carrée.

4°) Géométries des complexes contenant des liaisons pontées :
Dans certains complexes, on observe des molécules qui présentent des orbitales vides. Ces molécules se comportent comme acide de Lewis et reçoivent des doublets venant des ligands du complexe. Ces ligands forment ainsi entre ces molécules des ponts simples ou multiples.
a) Pont simple :
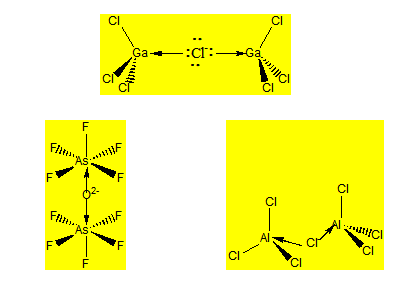
Ce sont les composés dans lesquels deux molécules se partagent un ligand. On peut avoir deux octaèdres ou tétraèdres accolés par un sommet.
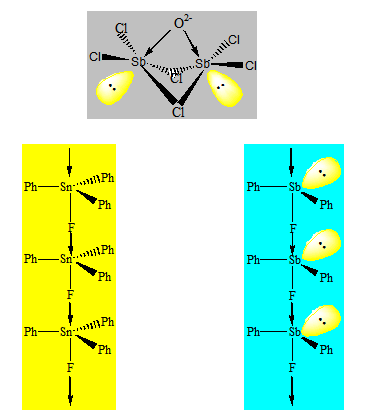
Considérons l’heptachlorodialuminate Al2Cl7- ou l’heptachlorodigallate Ga2Cl7- . Pour déterminer la géométrie de ce dernier on dissocie cet anion en Cl- et 2 GaCl3. GaCl3 est une molécule plane triangulaire et c’est un acide de Lewis par rapport à Ga. Cl- est une base de Lewis donc sera partagée entre deux GaCl3. L’environnement de Ga passe d’un environnement triangulaire à un environnement tétraédrique. La géométrie est donc constituée de deux tétraèdres accolés par un sommet. L’angle du pont est voisin de 109° puisqu’il y a en tout quatre paires libres.
b) Pont double :
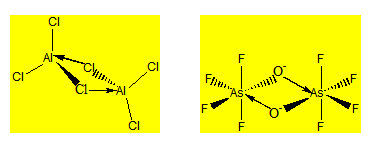
Ces composés sont formés par deux molécules qui se comportent comme acide de Lewis par rapport à l’atome central et comme base de Lewis par rapport aux atomes liants. On peut avoir deux tétraèdres ou deux octaèdres accolés par un côté.
Le trichlorure d’aluminium à l’état de vapeur est dimère et sa formule est Al2Cl6. Pour rendre compte de sa géométrie on considère que les deux halogénures d’aluminium s’associent par liaison dative. On a deux tétraèdres accolés par un côté.
c) Pont triple :
Dans ces composés il existe deux molécules qui se partagent d’une part un atome et d’autre part chacune d’elle se comporte comme acide de Lewis par rapport à l’atome central et comme base de Lewis par rapport aux atomes liants. On peut avoir deux pyramides à base carrée accolés par un côté et un sommet (ou une face). On peut également avoir deux octaèdres accolés par un côté et un sommet (face). On peut aussi rencontrer des complexes qui ont des structures en chaîne ou en couches infinies.
Considérons le nonachlorodithallate Tl2Cl93- qu’on dissocie en 3 Cl- et 2 TlCl3 qui sont des acides de Lewis. Nous pouvons avoir deux possibilités :
- Chaque TlCl3 reçoit un Cl- et les deux TlCl3 se partagent un Cl- ;
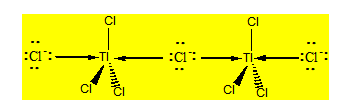
- Les deux TlCl3 se partagent 3Cl- suivant le même principe que le pont simple.
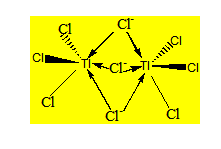
Cette dernière géométrie est la seule qui est stable ; l’autre bien qu’étant théoriquement possible n’a jamais été isolée.
5°) Liaisons multi centrées :
On appelle liaison multi centrée une liaison dans laquelle plusieurs atomes se partagent une seule paire électronique. Ces types de liaisons sont surtout rencontrés dans les molécules à déficience électronique comme les boranes, les carboranes, etc…. On ne fait intervenir ces liaisons que lorsque les composés sont déficients en électrons.
a) Liaison banane :
La réduction des halogénures de bore peut conduire à des boranes BH3 :
BCl3 + 3 NaH → BH3 + 3 NaCl.
Cependant le monomère BH3 n’a jamais pu être isolé et toutes les synthèses mènent au diborane B2H6.
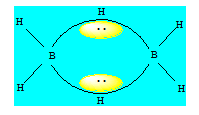
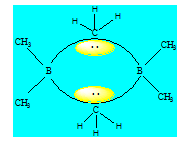
Ce type de liaison contient une paire électronique délocalisée sur trois atomes créant ainsi dans la couche de valence de chacun de ces atomes une densité faible par rapport à une liaison normale. B2H6 est composé de deux molécules BH3 séparées par des ponts hydrogènes. Pour rendre compte des liaisons dans la molécule, on considère que chaque bore est hybridé sp3.
- Les liaisons simples entre le bore et l’hydrogène proviennent d’un recouvrement de type axial entre une orbitale 1s de l’hydrogène et une orbitale sp3 du bore ;
- Les liaisons du pont entre le bore et l’hydrogène proviennent d’un recouvrement de type axial entre une orbitale 1s de l’hydrogène et deux orbitales sp3 du bore dans lesquelles l’une contient un électron célibataire et l’autre est vide. La paire électronique du pont est délocalisée sur les trois atomes (B – H – B) ; deux électrons reliant 3 cœurs ; la paire occupe le centre de gravité des trois sommets du triangle formé par les trois atomes.
Le remplacement d’un atome de bore dans un borane par un atome de carbone donne un carborane. Le composé C2B10H12 est isoélectronique à B12H122-.
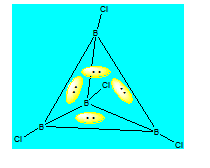
Quand une paire relie 4 cœurs ; ces derniers sont au sommet d’un tétraèdre. C’est l’exemple de B4Cl4. Il en résulte au niveau du pont une liaison tricentre fermée. Avec ce genre de liaison on peut construire plusieurs composés du bore dont le plus simple est le tétrahydroborate ou hydrure de bore BH4-.
b) Carbone à cinq pattes :
Ce sont des composés fabriqués à partir des boranes ou carboranes avec substitution de l’hydrogène par un groupe alkyle ; on obtient en général des composés ayant des atomes de carbone à 5 pattes.
Si dans une liaison banane le pont est constitué par un atome de carbone alors sa valence formelle est de cinq d’où le nom de carbone à cinq pattes.
c) Structures de type P4O10 et P4O6 :
Chaque fois qu’on a une molécule de type M4X6, la structure est toujours une cage tétraédrique Si on a M4X7, M4X8, M4X9, M4X10 on aura une structure tétraédrique à laquelle on ajoute 1, 2, 3 ou 4X.