Termes spectroscopiques
1°) Termes de Russell – Saunders Si on considère un atome polyélectronique le moment magnétique orbital de l’atome sera obtenu en faisant la somme de la contribution des différents électrons. De même le moment magnétique total de spin sera obtenu en considérant la somme des contributions de chacun des électrons. Chaque électron a un moment magnétique angulaire orbital individuel et un moment de spin individuel. Ces moments peuvent se coupler de deux façons. 1er cas : Les moments magnétiques individuels d’orbitales se couplent fortement entre eux pour donner une résultante notée . Les moments magnétiques de spin des différents électrons se couplent fortement entre eux pour donner une résultante notée . Les deux résultantes et se couplent faiblement pour donner un moment magnétique total noté . On appellera ceci le couplage de Russell – Saunders. 2ème cas : Les moments individuels d’orbitales et de spin et se couplent très fortement pour donner un moment angulaire total ensuite ces différents moments individuels totaux se couplent faiblement pour donner un moment angulaire total . On appelle ceci un couplage spin – orbite. Il a été montré expérimentalement que l’interaction et est d’autant plus faible que les atomes sont légers. Ainsi pour les éléments légers nous utiliserons principalement le 1er cas. Quand les éléments sont lourds c’est – à – dire au-delà du chrome il faut nécessairement faire intervenir le couplage spin – orbite. Pour définir une orbitale il faut avoir la valeur de l de la sous – couche à laquelle il appartient et la valeur du nombre quantique magnétique m ; mais dans chaque sous – couche la valeur de l est toujours égale au maximum de m. On peut déterminer les valeurs possibles des nombres quantiques L et S sachant que pour chaque valeur de L, le nombre quantique ML peut prendre les 2L+1 valeurs L, L-1, …,-L+1, -L et que pour chaque valeur de S, le nombre quantique MS peut prendre les 2S+1 valeurs +S, S-1,…,-S+1,-S. Dans le cas des atomes on sait que l’énergie des sous – couches est liée à la valeur de l (nombre quantique secondaire). Rappelons que pour n donné, l peut prendre les valeurs entières de 0 à n – 1. Pour l donné le nombre quantique magnétique m prend les valeurs de – l à + l. Une fois qu’on aura déterminé l’ensemble des valeurs de ML et de MS il suffira de combiner à chacune des valeurs de ML une valeur de MS. On définit ainsi l’ensemble des combinaisons possibles appelé micro – états : toutes les façons possibles de placer les électrons d’un métal dans les orbitales. A partir de ces derniers on définira les valeurs de et de qui permettent d’obtenir les termes de Russell – Saunders. Chaque terme est représenté par la notation 2S+1Γ où 2S+1 est la multiplicité de spin et Γ est mis pour S, P, D, F, G,… selon que L vaut 0, 1, 2, 3, 4,… Exemples : Li : 1s22s1 ; ML = 0 d’où L = 0 on a une orbitale de type S. MS = ½ d’où 2S. Be : 1s22s2 ; ML = 0 et MS = 0 d’où 1S. C : 1s22s22p2; la statistique permet d'évaluer le nombre de possibilités de ranger p objets dans n cases, selon : C’est le nombre de micro – états où n est le nombre maximal d’électrons dans la sous – couche et p le nombre d’électrons dans la sous - couche. Sous – couche p : 2 électrons ; le nombre de micro – états est +1 0 -1 ↑ ↑ ML = 1 + 0 = 1 ; MS = ½ + ½ = 1 ↑ ↑ ML = 1 - 1 = 0 ; MS = ½ + ½ = 1 ↑ ↑ ML = 0 - 1 = 1 ; MS = ½ + ½ = 1 ↓ ↓ ML = - 1 + 0 = - 1 ; MS = - ½ - ½ = - 1 ↓ ↓ ML = 1 - 1 = 0 ; MS = - ½ - ½ = - 1 ↓ ↓ ML = 0 - 1 = -1 ; MS = - ½ - ½ = - 1 ↓ ↑ ML = 1 + 0 = 1 ; MS = - ½ + ½ = 0 ↓ ↑ ML = 1 - 1 = 0 ; MS = - ½ + ½ = 0 ↓ ↑ ML = 0 - 1 = - 1 ; MS = - ½ + ½ = 0 ↑ ↓ ML = 1 + 0 = 1 ; MS = - ½ + ½ = 0 ↑ ↓ ML = 1 - 1 = 0 ; MS = - ½ + ½ = 0 ↑ ↓ ML = 0 - 1 = -1 ; MS = ½ - ½ = 0 ↑↓ ML = 1 + (1) = 2 ; MS = ½ - ½ = 0 ↑↓ ML = 0 + 0 = 0 ; MS = ½ - ½ = 0 ↑↓ ML= -1 - 1 = -2 ; MS = ½ - ½ = 0 Les 15 micro - états sont les suivants : ML MS = 1 MS = 0 MS = -1 2 - (1+,1-) - 1 (1+,0+) (1+,0-),(1-,0+) (1-,0-) 0 (1-,-1+) (1+,-1-),(1-,-1+),(0+,0-) (1-,-1-) -1 (0+,-1+) (0+,-1-),(0-,-1+) (0-,-1-) -2 - (-1+,-1-) - En résumé on a : ML MS +2 ↑↓ 0 0 ↑↓ 0 -2 ↑↓ 0 +1 ↑ ↑ +1 0 ↑ ↑ +1 -1 ↑ ↑ +1 +1 ↓ ↓ -1 0 ↓ ↓ -1 -1 ↓ ↓ -1 +1 ↓ ↑ 0 0 ↓ ↑ 0 -1 ↓ ↑ 0 +1 ↑ ↓ 0 0 ↑ ↓ 0 -1 ↑ ↓ 0 Prenons le micro – état (1+,1-) qui correspond à ML = 2 d’où le terme est D et MS = 0 d’où 1D. ML = 1 d’où le terme est P ; MS = 1 d’où 3P. ML = 0 d’où le terme est S et MS = 0 d’où 1S. Finalement les termes du carbone sont : 1S1D3P. Lequel correspond au terme fondamental 3P. Quand un atome a plusieurs termes ; le terme de Russell – Saunders de l’état fondamental est celui qui correspond à la multiplicité 2MS + 1 des spins, maximal. Le terme de l’état fondamental est donc 3P. Les termes S et D ont la même multiplicité du spin c’est - à – dire 1. La règle de Hund stipule que lorsque 2 termes de Russell – Saunders ont la même multiplicité du spin le terme de plus basse énergie est celui qui correspond à la valeur de L la plus grande donc 1D et enfin 1S. Les termes de Russell – Saunders du carbone sont : 3P1D1S. Pour un d2 on a : +2 +1 0 - 1 - 2 ↑↓ = 4 +2 +1 0 - 1 - 2 ↑ ↑ = ½ + ½ = 1 lorsque les électrons sont découplés. Les 45 micro - états classés de ML et MS sont rassemblés au tableau suivant : Les micro - états d’un ion d2 ML MS = 1 MS = 0 MS = -1 4 (2+,2-) 3 (2+,1+) (2+,1-),(2-,1+) (2-,1-) 2 (2+,0+) (2+,0-),(2-,0+),(1+,1-) (2-,0-) 1 (2+,-1+),(1+,0+) (2+,-1-),(2-,-1+),(1+,0-),(1-,0+) (2-,-1-),(1-,0-) 0 (2+,-2+),(1-,-1+) (2+,-2-),(2-,-2+),(1+,-1-),(1-,-1+),(0+,0-) (2-,-2-),(1-,-1-) -1 (1+,-2+),(0+,-1+) (1+,-2-),(1-,-2+),(0+,-1-),(0-,-1+) (1-,-2-),(0-,-1-) -2 (0+,-2+) (0+,-2-),(0-,-2+),(-1+,-1-) (0-,-2-) -3 (-1+,-2+) (-1+,-2-),(-1-,-2+) (-1-,-2-) -4 (-2+,-2-) Les termes de Russell – Saunders des orbitales s et p Configuration Dégénérescence Termes s1 2S s2 1S p1 et p5 6 2P p2 et p4 15 1S, 1D, 3P p3 20 2P, 2D, 4S Les termes de Russell – Saunders des orbitales d (Termes fondamentaux en gras Configuration Dégénérescence Termes d0 et d10 1S d1 et d9 10 2D d2 et d8 45 3F, 1G, 1D, 3P, 1S d3 et d7 120 4F, 4P, 2G, 2H, 2P, 2D, 2F, 2D d4 et d6 210 5D,3H, 3G, 3F, 3D, 3P, 3P, 1I, 1G, 1G, 1F, 1D, 1D, 1S, 1S d5 252 6S, 4G, 4F, 4D, 4P, 2I, 2H, 2G, 2G, 2F, 2F, 2D, 2D, 2D, 2P, 2S Un couplage spin – orbite se produit si L et S s’associent. On définit le paramètre d’interaction J par la relation : J = L + S. Ce couplage non seulement lève partiellement la dégénérescence de chaque terme mais peut aussi coupler les termes n’ayant ni les mêmes symétries orbitales et ni les mêmes multiplicités de spin en mélangeant les états ayant le même J. 2°) Application du couplage aux spectres électroniques Les termes de Russell – Saunders dans un champ octaédrique éclatent en plusieurs composantes. Les transitions qui peuvent intervenir ne peuvent se faire qu’entre des termes de même multiplicité de spin parce que le nombre d’électrons célibataires doit rester le même. C’est pour cela que des composés tels que [Mn(H2O)6]2+ qui contiennent un métal en d5 devraient en principe être incolores tout comme les complexes du zinc. La couleur d’un complexe est déterminée par les longueurs d’onde non absorbées par ce complexe. Dans le cas de [Mn(H2O)6]2+ il existe la levée d’interdiction c’est – à – dire le saut n’est pas permis et s’il peut se faire ce sera avec beaucoup de difficultés. Tout se passe comme si l’électron quitte l’état fondamental et retombe à cet état sans arriver à l’état d’arrivée. Ce saut interdit est permis par les vibrations de la molécule ; pour cela les bandes de ces sauts interdits sont extrêmement faibles. Les termes éclatent en symétrie Oh de la manière suivante : S → A1g P → T1g D → Eg + T2g F → A2g + T1g + T2g G → A1g + Eg + T1g + T2g H → Eg + 2T1g + T2g I → A1g + A2g + Eg + T1g + 2T2g Les composantes peuvent être une fois dégénérées (espèces A), doublement dégénérées (espèces E) ou triplement dégénérées (espèces T). Les indices 1 et 2 pour distinguer les états de dégénérescence semblables. Exemple : [Ti(H2O)6]3+ a une bande ; Ti3+ (d1) a pour termes de Russell – Saunders 2D. Les composantes dardent la multiplicité du terme ; 2D → 2Eg + 2T2g. Transition : T2g → Eg On voit qu’il y a 3 façons d’écrire l’état fondamental et que l’une des composantes de Russell – Saunders de l’état fondamental a une dégénérescence d’ordre 3. On peut écrire l’état d’arrivée de 2 façons différentes. L’une des composantes des termes de Russell – Saunders de l’état d’arrivée a une dégénérescence d’ordre 2. C’est donc le nombre de fois qu’il est possible d’écrire l’état fondamental en l’état d’arrivée qui permettra sur la base de la dégénérescence laquelle des composantes est la composante fondamentale du terme fondamental de Russell – Saunders. La ou les autres composantes sont les composantes d’arrivée. Il y a qu’une seule composante fondamentale et plusieurs composantes d’arrivée. Au lieu de dire que la bande du spectre de [Ti(H2O)6]3+ est dû à une transition des orbitales t2g vers les orbitales eg on dira que cette transition se fait d’un état T2g à un état Eg. 3°) Diagramme d’Orgel Soit le complexe [Cu(H2O)6]2+; Cu2+ ; 3d9 → 2D Transition Eg → T2g. Si on associe à un métal donné des ligands de plus en plus champ fort on constate une différence de plus en plus grande entre T2g et Eg. Si on considère Cu2+ qui est d9 on a un diagramme inverse par rapport à celui de Ti3+ qui est d1. Un tel diagramme représentant l’énergie en fonction des composantes est appelé diagramme d’Orgel. Maintenant pour le diagramme d’un d1 et d9 tétraédrique 2D → 2E + 2T2. Donc pour un d1 ; 2E → 2T2. Pour un d9 on a : Donc pour un d9 ; 2T2 → 2E. Finalement : et Quand 2 métaux ont sur leur sous – couche d des nombres d’électrons complémentaires le diagramme d’Orgel de l’un en configuration tétraédrique est l’équivalent du diagramme d’Orgel de l’autre en configuration octaédrique ; alors que le diagramme d’Orgel de l’un en configuration tétraédrique ou octaédrique est l’inverse du diagramme d’Orgel de l’autre en configuration tétraédrique ou octaédrique. Les diagrammes d’Orgel permettent de rendre compte des spectres des complexes de type « high spin ». Pour les complexes de type « low spin » on fait intervenir les diagrammes de Tanabé – Sugano. 4°) Diagramme de Tanabé - Sugano Il n’est plus possible dans le cas des complexes de type « low spin » d’appliquer la règle de Laporte ou règle des électrons célibataires (les transitions entre les niveaux énergétiques provenant de la même sous – couche d sont interdites). Il faudra raisonner sur les termes de Russell – Saunders correspondant à l’état fondamental en considérant un complexe « low spin ». Soit l’exemple de [Co(NH3)6]3+ qui est un complexe diamagnétique. Les termes de Russell – Saunders de Co3+ sont : 5D 3H 3F 3G 1I 1G 1S. Si on considère le diagramme de Tanabé – Sugano d’un d6 on voit qu’il contient le diagramme d’Orgel d’un d6 « high spin ». C’est la première partie de ce diagramme. On voit en fait que le terme de Russell – Saunders à l’état fondamental dans le cas d’un « high spin » est 5D qui éclate en 5T2g et 5Eg. Ceci permet de rendre compte de la bande unique des spectres d’un complexe « high spin » d’un d6. Quand le complexe est « low spin » dans le cas d’un d6 le terme de Russell – Saunders à l’état fondamental n’est plus 5D mais 1I. Si on considère le remplissage des orbitales t2g dans un environnement Oh on voit qu’il n’y a qu’une seule façon d’écrire cet arrangement c’est – à – dire : C’est pour cela que la composante fondamentale est A1g. Et l’ensemble des transitions partiront de cette composante fondamentale vers les autres composantes dérivant des termes de Russell – Saunders de multiplicité 1. En réalité le spectre de [Co(NH3)6]3+ comporte 2 bandes. Si on considère une première transition simple on obtient les configurations : La configuration 1 est triplement dégénérée on l’appelle T1g. Si on considère un double saut électronique on obtient 2 qui est triplement dégénérée et qu’on appellera T2g. Les 2 absorptions présentes sur le spectre de [Co(NH3)6]3+ correspondant bien aux transitions : A1g → T1g ; A1g → T2g. Les autres transitions bien que possibles théoriquement ne peuvent pas être décelées par le spectrophotomètre U.V. 5°) Couplages spin – orbite Pour trouver les niveaux d'énergie issus de l’action du couplage spin - orbite sur un terme donné, il faut donc calculer toutes les valeurs possibles du nombre quantique MJ associé à et en déduire les valeurs possibles de J sachant que pour chaque valeur de J, MJ prend les 2J+1 valeurs J, J-1, . . .,-J + 1,-J. Le niveau obtenu est noté en ajoutant au terme dont il est issu l’indice J. Par exemple pour le terme fondamental 3F de la configuration d2, nous avons: S = 1, d'où MS = ±1, 0 L = 3, d'où ML = ± 3, ± 2, ±1, 0 On en déduit que les valeurs possibles de J sont : J = 4 auquel correspond MJ = ± 4, ± 3, ± 2, ±1, 0 J = 3 auquel correspond MJ = ± 3, ± 2, ±1, 0 J = 2 auquel correspond MJ = ± 2, ±1, 0 Les niveaux issus de 3F auxquels on donnera le nom d'état seront notés 3F4, 3F3 et 3F2. La dégénérescence 2J +1 de chacun de ces états ne peut être complètement levée que par l’action d'une perturbation externe, par exemple un champ magnétique. Une règle empirique proposée à nouveau par Hund permet de prévoir l’état fondamental. Si la couche est moins que demi - remplie (configuration d1 à d4), l’état fondamental est donné par la plus petite valeur de J; si la couche est plus que demi - remplie, c’est la plus grande valeur de J qui correspond à l’état fondamental. Cet état fondamental, dans le cas de l’ion d2, est donc3F2. Le couplage spin - orbite non seulement lève partiellement la dégénérescence de chaque terme mais peut aussi coupler des termes n’ayant ni les mêmes symétries orbitales et ni les mêmes multiplicités de spin en mélangeant les états ayant le même J. Cet effet du second ordre est d’autant plus important que le couplage spin - orbite est plus fort et que les termes sont moins espacés, c’est-à-dire que la répulsion électronique est faible. Ce couplage de différents termes est ainsi à peu près négligeable pour les ions de transition de la première série, il joue un certain rôle avec ceux de la seconde série et plus encore de la troisième. A titre d’exemple, nous représentons les effets successifs des différentes perturbations dans le cas d’une configuration d1, il n’intervient alors que le couplage spin - orbite, et d’une configuration d2. Bien que pour la clarté de la figure l’échelle des énergies n’ait pu être respectée, nous précisons les valeurs des énergies à partir de l’état fondamental obtenues expérimentalement pour les ions Ti3+ (d1) et V3+ (d2) et cela afin de bien mettre en évidence les importances relatives de la répulsion électronique et du couplage spin - orbite. Niveaux d'énergie pour l'ion libre Ti3+ de configuration d1 Niveaux d'énergie pour l'ion libre V3+ de configuration d2 6º) Propriétés optiques des composés de coordination (transitions d-d et interprétation qualitative des spectres) La couleur des complexes résulte de transitions intraconfigurationnelles entre orbitales d. En effet, la différence d'énergie entre orbitales t2g et eg égale à Δ varie entre 12 500 cm-1 et 25 000 cm-1, c'est-à-dire une gamme d'énergie correspondant au spectre visible du rayonnement électromagnétique (800 nm > λ > 400 nm). Une transition électronique correspond au passage d'un électron du niveau fondamental vers un niveau excité. Lorsqu'elle a lieu, la matière absorbe un photon dont l'énergie correspond rigoureusement à la différence d'énergie entre le terme fondamental et un terme excité. Elle suit également des règles de sélection : l'une liée au moment orbital et l'autre lié au moment de spin. Une transition autorisée a lieu si les orbitales impliquées au cours de ce processus sont telles que Δl = ± 1 et ΔS = 0. Les transitions d - d sont interdites selon la première règle car Δl = 0, mais elles sont observées grâce à des mécanismes de relaxation liés essentiellement à des couplages électrons-noyaux (rôle des vibrations). Par contre une transition d - d avec changement de spin est interdite selon les deux règles. Là encore, elle peut être observée grâce à des mécanismes de relaxation beaucoup moins efficaces liés aux couplages entre moments de spin et moments orbitaux des électrons. Par conséquent, ces transitions sont beaucoup moins intenses que les transitions d - d permises de spin. Nous ne parlerons que des transitions permises de spin dans la suite. Le spectre électronique (mesure de la variation de l'absorbance d'une solution en fonction de λ) peut être formé d'une ou de plusieurs transitions électroniques. Si un complexe ne présente qu'une seule transition, alors la couleur de ce complexe correspond à la couleur complémentaire de la couleur absorbée pendant la transition. 6-1 Spectres à une bande : interprétation de la couleur des complexes Cette situation se produit dans les complexes des ions Ti3+ de configuration d1. Une solution aqueuse de [Ti(H2O)6]3+ est rouge. Le spectre d'absorption est formé d'une seule bande avec λmax = 510 nm. La couleur absorbée est le vert, le complexe apparaît de la couleur complémentaire à savoir le rouge. La bande observée est interprétée comme la transition de l'électron de l'orbitale t2g vers l'orbitale eg. L'énergie de cette transition est égale à Δ. On obtient donc la valeur de Δ à 510 nm, soit en unités cm-1, 19 607 cm-1. Un cas similaire est celui de l'ion hexa-aquo de cuivre (II) : [Cu(H2O)6]2+ de configuration d9. Dans ce cas, le composé est bleu-vert, et son spectre est constitué d'une seule bande située à 840 nm. Il absorbe le rouge, et apparaît donc de la couleur complémentaire. Le spectre s'analyse de façon semblable. On peut voir cette transition comme le passage de l'électron d'une orbitale t2g à une orbitale eg. La transition possède toujours l'énergie Δ. Dans ce cas, Δ vaut 12 000 cm-1 (λ = 833,3 nm). De l'analyse de ces deux spectres, on donne une preuve expérimentale que le champ cristallin Δ augmente avec la charge de l'ion de transition. De manière générale, ΔM3+ > ΔM2+. 6-2 Spectre à deux bandes : rôle de la répulsion « électron-électron » Examinons maintenant le spectre visible du complexe [Cr(H2O)6]3+ dont la configuration de plus basse énergie est t2g3 (d3). Celui-ci est formé de deux bandes d'intensité semblable à 17 400 cm-1 (λ = 575 nm) et 24 600 cm-1 (λ = 406,5 nm), dont l'origine est à chercher dans des transitions t2g → eg. Une façon de réaliser cette transition est de promouvoir l'électron de l'orbitale dxy vers l'orbitale dz2. Dans ce cas, la densité électronique est relocalisée au cours de la transition d'un plan xy dans une direction z déjà riche en électron (les orbitales dzx et dyz sont occupées). Mais il existe une autre façon de réaliser cette transition en faisant passer l'électron de l'orbitale dzx vers l'orbitale dz2. Dans ce cas, la densité électronique sera simplement relocalisée selon un axe z déjà riche en électron avant la transition. On conçoit aisément que pour le premier cas la répulsion électronique va beaucoup augmenter au cours de la transition, et beaucoup moins dans le second cas. Par conséquent les deux transitions envisagées apparaissent à des énergies différentes. Les autres possibilités de transitions t2g → eg seront similaires à l'un ou l'autre cas, si bien que le complexe possède deux bandes dans son spectre d'absorption. En le premier cas, la densité électronique se déplace fortement vers les ligands sur l'axe z. Alors qu'au deuxième cas, le déplacement est beaucoup plus faible. Une question vient à l'esprit immédiatement, quelle transition va permettre de déterminer Δ? Intuitivement, on choisira la transition de plus basse énergie, et on aura raison. On détermine donc Δ = 17 400 cm-1 (λ = 575 nm). 7°) Interprétation quantitative des spectres Pour comprendre l'origine de la deuxième transition pour le complexe [Cr(H2O)6]3+, il faut étudier plus en détail la structure électronique des ions de transition et tenir compte de la répulsion électron-électron. Jusqu'à présent celle-ci a été introduite dans le cas où elle est maximum, c'est-à-dire quand deux électrons occupent la même orbitale, sous la forme d'une quantité énergétique P. Lorsque P est beaucoup plus grand que Δ0, la structure électronique de l'ion considéré est profondément modifiée : le complexe à champ fort préfère associer tous ses électrons dans des orbitales t2g par souci de gain en énergie. Lorsque deux électrons occupent deux orbitales différentes orientées dans l'espace dans une direction commune, la répulsion électron-électron est plus faible mais elle existe. Elle aura pour effet de créer à partir d'un état dégénéré issu d'une configuration plusieurs niveaux d'énergies différentes. Une configuration donnée (t2g)a(eg)b donne naissance, à cause de la répulsion « électron-électron », à plusieurs niveaux d'énergie différents, représentés par un terme spectroscopique. Les transitions entre termes spectroscopiques ont des énergies couvrant le domaine du visible. Les spectres optiques des composés de coordination ne s'interprètent donc pas comme un saut électronique t2g → eg, mais comme une transition entre termes spectroscopiques (T2g → Eg). De la même manière que nous avons comparé P et Δ0 (= 10 Dq) jusqu'à présent, nous allons comparer la répulsion électron-électron traitée dans sa globalité (qui sera notée par la suite RE) et le champ cristallin. Commençons par examiner deux cas limite : la situation champ faible Δ0 << RE et la situation champ fort Δ0 >> RE. 7-1 Structure électronique des complexes à champ faible Le champ cristallin est faible, les effets de la RE dominent : Δ0 << RE. Nous considérons d'abord l'effet de la RE sur les niveaux d'énergie de l'ion libre. Les cinq orbitales d (ou si l'on tient compte du spin les dix spin-orbitales) sont toutes de même énergie. Pour la configuration d3, il faut placer trois électrons (ou trois objets) parmi dix spin-orbitales (ou dix cases). La statistique permet d'évaluer le nombre de possibilités de ranger p objets dans n cases, selon : Avec n, nombre de spin-orbitales et p, le nombre d'électrons. Pour d3, n = 10, p = 3, soit . Il existe 120 possibilités de ranger 3 électrons sur 10 spin-orbitales. La configuration d3 regroupe à elle seule 120 états d'énergie équivalente égale à trois fois l'énergie d'une orbitale d, soit 3.Ed. La RE va lever cette dégénérescence en créant huit niveaux d'énergie différents qui sont les termes spectroscopiques. L'énergie de chaque terme est obtenue grâce à un calcul sophistiqué où interviennent des intégrales biélectroniques quantifiant la RE. A titre d'exemple pour la configuration d3, on obtient le diagramme énergétique des termes spectroscopiques de la figure ci – contre. Considérons maintenant l'effet du champ cristallin sur les termes spectroscopiques de l'ion libre. Celui-ci n'agit que sur la partie orbitalaire (la multiplicité de spin ne sera pas modifiée) en continuant à lever la dégénérescence orbitale des termes. Par exemple, de la même façon que les cinq orbitales d sont éclatées sous l'effet d'un champ octaédrique en trois orbitales t2g et deux orbitales eg, les termes de symétrie D seront éclatés en termes T2g et Eg. Les termes de symétrie F seront transformés en . 7-2 Structure électronique des complexes à champ fort Δ0 >> RE, le champ cristallin est fort, et ses effets sur les niveaux d'énergie dominent. L'effet du champ cristallin sur une configuration dn est traité dans un premier temps. Ainsi les orbitales d deviennent en champ octaédrique les orbitales t2g d'énergie -2/5 Δ0 et les orbitales eg d'énergie -3/5 Δ0. A partir de la configuration d3, on obtient les configurations d'énergie et de dégénérescence différente : (t2g)3 d'énergie égale à 3(-2/5 Δ0) = -6/5 Δ0, (t2g)2(eg)1 d'énergie égale à 2(-2/5)Δ0 + 3/5Δ0 = - 1/5 Δ0, (t2g)1(eg)2 d'énergie égale à -2/5Δ0 + 2(3/5Δ0) = 4/5 Δ0, (eg)3 d'énergie égale à 3(3/5Δ0) = 9/5 Δ0. La RE a pour effet de lever partiellement cette dégénérescence pour créer les séries de termes spectroscopiques déjà identifiés dans la première situation. Cette situation donne accès à l'énergie des termes. 7-3 Corrélation « champ faible-champ fort ». Diagrammes de Tanabé-Sugano La réalité étant très souvent intermédiaire entre un champ faible et un champ fort, on établit une corrélation termes à termes de l'approche champ faible vers l'approche champ fort (figure suivante). Termes spectroscopiques de l'ion complexé d3 champ faible et champ fort. Sous chaque terme est indiquée en rouge la dégénérescence associée à chaque niveau. Cette corrélation aboutit à la construction d'un diagramme de Tanabé-Sugano (figure ci - après), où l'énergie des termes est portée selon E/B = f (Δ/B), B (terme relatif à la répulsion électronique) étant le paramètre de Racah. Le terme fondamental est ainsi confondu avec l'axe des abscisses. Diagramme de Tanabé-Sugano pour la configuration d3 7-4 Interprétation Interpréter un spectre signifie que l'on est capable d'identifier les termes excités atteints lors de chacune des transitions en considérant le diagramme de Tanabé - Sugano décrivant la configuration étudiée. Une interprétation qualitative du spectre de [Cr(H2O)6]3+; (configuration d3, diagramme figure précédente) est la suivante : Le terme fondamental est 4A2g, terme issu de la configuration de plus basse énergie (t2g)3. La configuration excitée de plus basse énergie est (t2g)2(eg)1. La RE va créer deux termes spectroscopiques de même multiplicité de spin que le terme fondamental 4T2g et 4T1g. On obtient deux transitions dans le domaine du visible : 1ère transition : 4A2g → 4T2g ; 2ème transition : 4A2g → 4T1g ; A priori, une troisième transition devrait être observée, correspondant à la transition entre l'état fondamental 4A2g et le terme excité 4T1g le plus haut en énergie. Dans les faits, cette transition est trop haute en énergie pour être observée dans le domaine du visible. On remarque que l'on a volontairement négligé la transition vers les termes excités 2Eg, 2T1g. En effet la transition du terme fondamental 4A2g vers ces deux termes de même énergie est une transition qui modifie le spin du système. Ce type de transitions est interdit selon les règles de sélection, donc à priori non observable. Or, si on examine le spectre de manière attentive, il existe un épaulement dans le pied de la bande correspondant à la transition 4A2g → 4T2g ; situé en dessous de 16 000 cm-1 (λ = 625 nm). Cet épaulement correspond à la transition 4A2g → 4Eg, 2T1g peu intense masquée par la transition permise de spin. Des mécanismes de relaxation mentionnés au début de ce chapitre sont à l'origine de son observation. Une fois l'analyse qualitative effectuée, l'interprétation quantitative du spectre permet de déterminer Δ et B. Pour cela, nous allons effectuer une résolution graphique. Pour déterminer les deux inconnues Δ et B, nous allons effectuer une résolution graphique. Nous remarquons sur le diagramme que la pente du premier terme excité 4T2g est égale à un. Par conséquent, on a pour ce terme E2/B = 1. Δ/B, soit E2 = Δ. La première transition nous donne directement la valeur de Δ, soit Δ= 17400 cm-1. Pour estimer B, nous calculons le rapport (E2/B)/(E1/B) = E2/E1, soit 24600/17400 = 1,41. Cette astuce nous permet d'éliminer l'inconnue B sur un des axes. On cherche alors graphiquement le rapport correspondant E2/E1 = 1,41 sur l'axe des ordonnées, et quand il est trouvé, il suffit de lire sur l'axe des abscisses la valeur de Δ/B, soit dans ce cas 25. On trouve donc pour B = 700 cm-1. 8°) Propriétés magnétiques des composés de coordination 8-1 Description macroscopique et microscopique du magnétisme de composés moléculaires Une autre façon d'appréhender la force du champ cristallin est de mesurer les propriétés magnétiques des complexes. Deux types de comportements magnétiques sont attendus pour les complexes : le paramagnétisme (le composé est attiré vers la zone de champ magnétique maximum) et le diamagnétisme (le composé est repoussé vers la zone de champ magnétique minimum). Il s'agit de magnétisme induit, c'est-à-dire que la présence d'un champ magnétique extérieur est nécessaire pour mettre en évidence ces comportements. Il existe deux types de comportements magnétiques pour deux complexes de l'ion Fe2+ (configuration d6) : le complexe [Fe(H2O)6]2+ présent dans FeSO4.7H2O et le complexe K4[Fe(CN)6]. Dans le premier cas, il existe des électrons non appariés, le complexe est donc spin fort-champ faible. Dans le deuxième cas, les électrons sont tous appariés, le complexe est donc « spin faible-champ fort ». 8-2 Mesures magnétiques et loi de Curie Toute molécule résulte d’une association d’atomes, eux-mêmes constitués chacun d’un noyau chargé positivement autour duquel gravitent des électrons chargés négativement. Il existe une étroite relation entre la structure électronique et les propriétés magnétiques d’une molécule. L’étude des propriétés magnétiques permet de déterminer la structure des complexes des ions métalliques ainsi que la nature (anti ou ferromagnétique) de l’interaction entre deux ou plusieurs centres métalliques. Ainsi, il existe une étroite relation entre la structure et les propriétés magnétiques d’une molécule. Le magnétisme moléculaire est une technique qui permet d’appréhender la géométrie et la structure électronique des ions métalliques à partir de la susceptibilité magnétique. Pour étudier les propriétés magnétiques d’une substance, on la soumet à l’action d’un champ magnétique intense. L’échantillon acquiert alors une aimantation. En effet en présence d’un champ magnétique , les différents moments magnétiques électroniques ou nucléaires vont se diviser en différents niveaux d’énergie. Pour le noyau d’hydrogène caractérisé par un spin de valeur 1/2, l’aimantation peut prendre deux positions dites parallèle ou anti parallèle. L’état parallèle étant de plus basse énergie, il est plus peuplé et il en résulte dans le milieu, une aimantation nucléaire macroscopique notée M (moment magnétique par unité de volume). Cette aimantation est proportionnelle à l’intensité du champ magnétique appliqué. Le coefficient de proportionnalité, noté , définit la susceptibilité magnétique du milieu considéré : M = H. Lorsque est positif on dit que le corps dans lequel apparaît l’aimantation est paramagnétique; cet effet provient des électrons non appariés de la substance. Lorsqu’ il est négatif, le corps est dit diamagnétique ; la substance est légèrement repoussée par le champ magnétique, cet effet provient des électrons appariés. Le magnétisme moléculaire est donc une technique qui permet de déterminer l’état de perturbation d’un ion métallique à partir de la susceptibilité. La susceptibilité diamagnétique, présente dans tous les composés, est négative et indépendante de la température. Elle est liée à la circulation des électrons appariés dans leurs orbitales induite par le champ magnétique externe. Elle se calcule à partir de contributions atomiques constituant la substance étudiée. La susceptibilité paramagnétique existe uniquement pour les composés possédant des électrons non appariés. Elle est positive et dépendante de la température. Elle est beaucoup plus importante que la susceptibilité diamagnétique. Pour les complexes, il est utile de définir une susceptibilité molaire notée χM. Les susceptibilités diamagnétiques molaires par atome ou groupe d’atomes sont données au tableau suivant : Atomes i (106 cm3/mol) Atomes i (106 cm3/mol) H 2,85 O alcool, éther 4,61 C 6,00 O aldéhyde, cétone +1,66 N chaîne ouverte 5,55 O ester 3,36 N anneau 4,61 F 11,5 N monoacide 1,54 Cl 20,10 N diamide, imide 2,11 Br 30,6 Si 13 I 44,6 P 10 S 15 As 21 Se 23 La dépendance thermique de la susceptibilité paramagnétique molaire est donnée par la loi de Curie, selon la relation : (cm3mol-1) g est le facteur Zeeman du complexe étudié, et est proche du facteur gyromagnétique de l'électron ge = 2.0023. On appelle la Constante de Curie C la quantité égale à ; elle dépend du spin étudié et du facteur g du complexe étudié (figure suivante). On peut récrire la loi de Curie sous la forme d'un produit indépendant de la température χMT = C. S χMT = C (cm3mol-1K) 1/2 0,375 1 1 3/2 1,875 2 3 5/2 4,375 Par exemple, le complexe [FeII(H2O)6]2+ possède un spin s = 2 et g = 2, donc le produit χMT vaut 3 cm3mol-1K. Le complexe diamagnétique [FeII(CN)6]4- possède un spin s = 0, d'où χMT est nul. Le complexe [CrIII(H2O)6]3+ a trois électrons célibataires dans les orbitales t2g, donc s = 3/2, soit χMT = 1,875 cm3mol-1K avec g = 2. Ainsi, la valeur du produit χMT permet de connaître le nombre d'électrons célibataires dans un composé.
| Site: | Touch By SukaJanda01 |
| Cours: | CHIM 4211 : Chimie inorganique et des solides II |
| Livre: | Termes spectroscopiques |
| Imprimé par: | Visiteur anonyme |
| Date: | jeudi 3 juillet 2025, 15:27 |
Table des matières
- 1 Termes de Russell – Saunders
- 2 Application du couplage aux spectres électroniques
- 3 Diagramme d’Orgel
- 4 Diagramme de Tanabé - Sugano
- 5 Couplages spin – orbite
- 6 Propriétés optiques des composés de coordination (transitions d-d et interprétation qualitative des spectres)
- 7 Interprétation quantitative des spectres
- 8 Propriétés magnétiques des composés de coordination
1 Termes de Russell – Saunders
Si on considère un atome polyélectronique le moment magnétique orbital de l’atome sera obtenu en faisant la somme de la contribution des différents électrons. De même le moment magnétique total de spin sera obtenu en considérant la somme des contributions de chacun des électrons. Chaque électron a un moment magnétique angulaire orbital individuel et un moment de spin individuel. Ces moments peuvent se coupler de deux façons.
1er cas : Les moments magnétiques individuels d’orbitales se couplent fortement entre eux pour donner une résultante notée . Les moments magnétiques de spin des différents électrons se couplent fortement entre eux pour donner une résultante notée . Les deux résultantes et se couplent faiblement pour donner un moment magnétique total noté . On appellera ceci le couplage de Russell – Saunders.
2ème cas : Les moments individuels d’orbitales et de spin et se couplent très fortement pour donner un moment angulaire total ensuite ces différents moments individuels totaux se couplent faiblement pour donner un moment angulaire total . On appelle ceci un couplage spin – orbite. Il a été montré expérimentalement que l’interaction et est d’autant plus faible que les atomes sont légers. Ainsi pour les éléments légers nous utiliserons principalement le 1er cas. Quand les éléments sont lourds c’est – à – dire au-delà du chrome il faut nécessairement faire intervenir le couplage spin – orbite.
Pour définir une orbitale il faut avoir la valeur de l de la sous – couche à laquelle il appartient et la valeur du nombre quantique magnétique m ; mais dans chaque sous – couche la valeur de l est toujours égale au maximum de m.
On peut déterminer les valeurs possibles des nombres quantiques L et S sachant que pour chaque valeur de L, le nombre quantique ML peut prendre les 2L+1 valeurs L, L-1, …,-L+1, -L et que pour chaque valeur de S, le nombre quantique MS peut prendre les 2S+1 valeurs +S, S-1,…,-S+1,-S. Dans le cas des atomes on sait que l’énergie des sous – couches est liée à la valeur de l (nombre quantique secondaire). Rappelons que pour n donné, l peut prendre les valeurs entières de 0 à n – 1. Pour l donné le nombre quantique magnétique m prend les valeurs de – l à + l.
Une fois qu’on aura déterminé l’ensemble des valeurs de ML et de MS il suffira de combiner à chacune des valeurs de ML une valeur de MS. On définit ainsi l’ensemble des combinaisons possibles appelé micro – états : toutes les façons possibles de placer les électrons d’un métal dans les orbitales. A partir de ces derniers on définira les valeurs de et de qui permettent d’obtenir les termes de Russell – Saunders.
Chaque terme est représenté par la notation 2S+1Γ où 2S+1 est la multiplicité de spin et Γ est mis pour S, P, D, F, G,… selon que L vaut 0, 1, 2, 3, 4,…
Exemples :
Li : 1s22s1 ; ML = 0 d’où L = 0 on a une orbitale de type S.
MS = ½ d’où 2S.
Be : 1s22s2 ; ML = 0 et MS = 0 d’où 1S.
C : 1s22s22p2; la statistique permet d'évaluer le nombre de possibilités de ranger p objets dans n cases, selon :
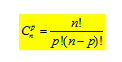
C’est le nombre de micro – états où n est le nombre maximal d’électrons dans la sous – couche et p le nombre d’électrons dans la sous - couche.
Sous – couche p : 2 électrons ; le nombre de micro – états est


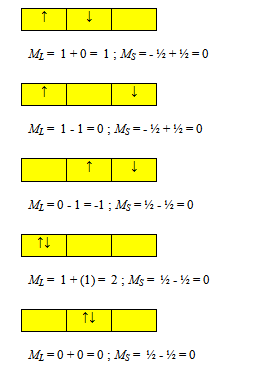
|
+1 |
0 |
-1 |
|
↑ |
↑ |
|
ML = 1 + 0 = 1 ; MS = ½ + ½ = 1
|
↑ |
|
↑ |
ML = 1 - 1 = 0 ; MS = ½ + ½ = 1
|
|
↑ |
↑ |
ML = 0 - 1 = 1 ; MS = ½ + ½ = 1
|
↓ |
↓ |
|
ML = - 1 + 0 = - 1 ; MS = - ½ - ½ = - 1
|
↓ |
|
↓ |
ML = 1 - 1 = 0 ; MS = - ½ - ½ = - 1
|
|
↓ |
↓ |
ML = 0 - 1 = -1 ; MS = - ½ - ½ = - 1
|
↓ |
↑ |
|
ML = 1 + 0 = 1 ; MS = - ½ + ½ = 0
|
↓ |
|
↑ |
ML = 1 - 1 = 0 ; MS = - ½ + ½ = 0
|
|
↓ |
↑ |
ML = 0 - 1 = - 1 ; MS = - ½ + ½ = 0
|
↑ |
↓ |
|
ML = 1 + 0 = 1 ; MS = - ½ + ½ = 0
|
↑ |
|
↓ |
ML = 1 - 1 = 0 ; MS = - ½ + ½ = 0
|
|
↑ |
↓ |
ML = 0 - 1 = -1 ; MS = ½ - ½ = 0
|
↑↓ |
|
|
ML = 1 + (1) = 2 ; MS = ½ - ½ = 0
|
|
↑↓ |
|
ML = 0 + 0 = 0 ; MS = ½ - ½ = 0
|
|
|
↑↓ |
ML= -1 - 1 = -2 ; MS = ½ - ½ = 0
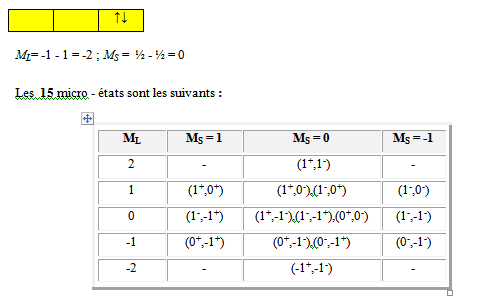
Les 15 micro - états sont les suivants :
|
ML |
MS = 1 |
MS = 0 |
MS = -1 |
|
2 |
- |
(1+,1-) |
- |
|
1 |
(1+,0+) |
(1+,0-),(1-,0+) |
(1-,0-) |
|
0 |
(1-,-1+) |
(1+,-1-),(1-,-1+),(0+,0-) |
(1-,-1-) |
|
-1 |
(0+,-1+) |
(0+,-1-),(0-,-1+) |
(0-,-1-) |
|
-2 |
- |
(-1+,-1-) |
- |
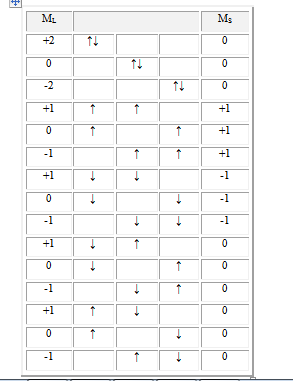
En résumé on a :
|
ML |
|
MS |
||
|
+2 |
↑↓ |
|
|
0 |
|
0 |
|
↑↓ |
|
0 |
|
-2 |
|
|
↑↓ |
0 |
|
+1 |
↑ |
↑ |
|
+1 |
|
0 |
↑ |
|
↑ |
+1 |
|
-1 |
|
↑ |
↑ |
+1 |
|
+1 |
↓ |
↓ |
|
-1 |
|
0 |
↓ |
|
↓ |
-1 |
|
-1 |
|
↓ |
↓ |
-1 |
|
+1 |
↓ |
↑ |
|
0 |
|
0 |
↓ |
|
↑ |
0 |
|
-1 |
|
↓ |
↑ |
0 |
|
+1 |
↑ |
↓ |
|
0 |
|
0 |
↑ |
|
↓ |
0 |
|
-1 |
|
↑ |
↓ |
0 |
Prenons le micro – état (1+,1-) qui correspond à ML = 2 d’où le terme est D et MS = 0 d’où 1D.
ML = 1 d’où le terme est P ; MS = 1 d’où 3P.
ML = 0 d’où le terme est S et MS = 0 d’où 1S.
Finalement les termes du carbone sont : 1S1D3P.
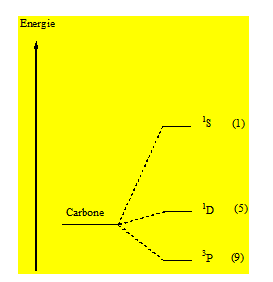
Lequel correspond au terme fondamental 3P.
Quand un atome a plusieurs termes ; le terme de Russell – Saunders de l’état fondamental est celui qui correspond à la multiplicité 2MS + 1 des spins, maximal. Le terme de l’état fondamental est donc 3P. Les termes S et D ont la même multiplicité du spin c’est - à – dire 1. La règle de Hund stipule que lorsque 2 termes de Russell – Saunders ont la même multiplicité du spin le terme de plus basse énergie est celui qui correspond à la valeur de L la plus grande donc 1D et enfin 1S. Les termes de Russell – Saunders du carbone sont : 3P1D1S.
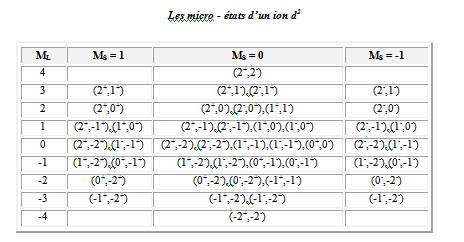
Pour un d2 on a :
+2 +1 0 - 1 - 2
|
↑↓ |
|
|
|
|
= 4
+2 +1 0 - 1 - 2
|
↑ |
↑ |
|
|
|
= ½ + ½ = 1 lorsque les électrons sont découplés.
Les 45 micro - états classés de ML et MS sont rassemblés au tableau suivant :
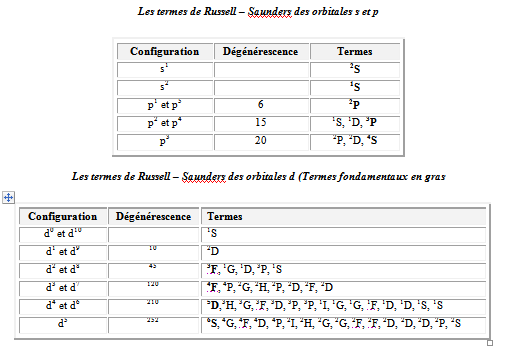
Les termes de Russell – Saunders des orbitales s et p
|
Configuration |
Dégénérescence |
Termes |
|
s1 |
|
2S |
|
s2 |
|
1S |
|
p1 et p5 |
6 |
2P |
|
p2 et p4 |
15 |
1S, 1D, 3P |
|
p3 |
20 |
2P, 2D, 4S |
Les termes de Russell – Saunders des orbitales d (Termes fondamentaux en gras
|
Configuration |
Dégénérescence |
Termes |
|
d0 et d10 |
|
1S |
|
d1 et d9 |
10 |
2D |
|
d2 et d8 |
45 |
3F, 1G, 1D, 3P, 1S |
|
d3 et d7 |
120 |
4F, 4P, 2G, 2H, 2P, 2D, 2F, 2D |
|
d4 et d6 |
210 |
5D,3H, 3G, 3F, 3D, 3P, 3P, 1I, 1G, 1G, 1F, 1D, 1D, 1S, 1S |
|
d5 |
252 |
6S, 4G, 4F, 4D, 4P, 2I, 2H, 2G, 2G, 2F, 2F, 2D, 2D, 2D, 2P, 2S |
Un couplage spin – orbite se produit si L et S s’associent. On définit le paramètre d’interaction J par la relation : J = L + S. Ce couplage non seulement lève partiellement la dégénérescence de chaque terme mais peut aussi coupler les termes n’ayant ni les mêmes symétries orbitales et ni les mêmes multiplicités de spin en mélangeant les états ayant le même J.
2 Application du couplage aux spectres électroniques
Les termes de Russell – Saunders dans un champ octaédrique éclatent en plusieurs composantes. Les transitions qui peuvent intervenir ne peuvent se faire qu’entre des termes de même multiplicité de spin parce que le nombre d’électrons célibataires doit rester le même. C’est pour cela que des composés tels que [Mn(H2O)6]2+ qui contiennent un métal en d5 devraient en principe être incolores tout comme les complexes du zinc. La couleur d’un complexe est déterminée par les longueurs d’onde non absorbées par ce complexe. Dans le cas de [Mn(H2O)6]2+ il existe la levée d’interdiction c’est – à – dire le saut n’est pas permis et s’il peut se faire ce sera avec beaucoup de difficultés. Tout se passe comme si l’électron quitte l’état fondamental et retombe à cet état sans arriver à l’état d’arrivée. Ce saut interdit est permis par les vibrations de la molécule ; pour cela les bandes de ces sauts interdits sont extrêmement faibles.
Les termes éclatent en symétrie Oh de la manière suivante :
S → A1g
P → T1g
D → Eg + T2g
F → A2g + T1g + T2g
G → A1g + Eg + T1g + T2g
H → Eg + 2T1g + T2g
I → A1g + A2g + Eg + T1g + 2T2g
Les composantes peuvent être une fois dégénérées (espèces A), doublement dégénérées (espèces E) ou triplement dégénérées (espèces T). Les indices 1 et 2 pour distinguer les états de dégénérescence semblables.
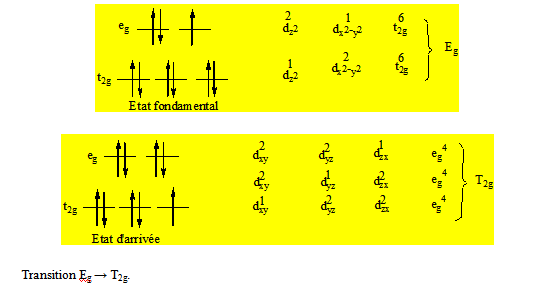
Exemple : [Ti(H2O)6]3+ a une bande ; Ti3+ (d1) a pour termes de Russell – Saunders 2D. Les composantes dardent la multiplicité du terme ; 2D → 2Eg + 2T2g.
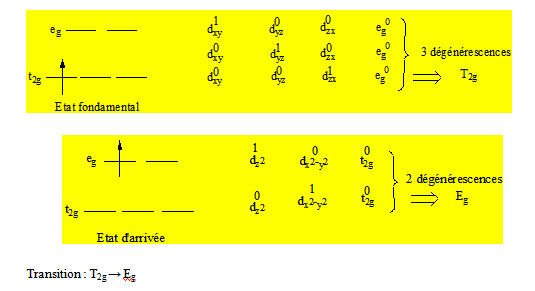
On voit qu’il y a 3 façons d’écrire l’état fondamental et que l’une des composantes de Russell – Saunders de l’état fondamental a une dégénérescence d’ordre 3. On peut écrire l’état d’arrivée de 2 façons différentes. L’une des composantes des termes de Russell – Saunders de l’état d’arrivée a une dégénérescence d’ordre 2. C’est donc le nombre de fois qu’il est possible d’écrire l’état fondamental en l’état d’arrivée qui permettra sur la base de la dégénérescence laquelle des composantes est la composante fondamentale du terme fondamental de Russell – Saunders. La ou les autres composantes sont les composantes d’arrivée. Il y a qu’une seule composante fondamentale et plusieurs composantes d’arrivée. Au lieu de dire que la bande du spectre de [Ti(H2O)6]3+ est dû à une transition des orbitales t2g vers les orbitales eg on dira que cette transition se fait d’un état T2g à un état Eg.
3 Diagramme d’Orgel
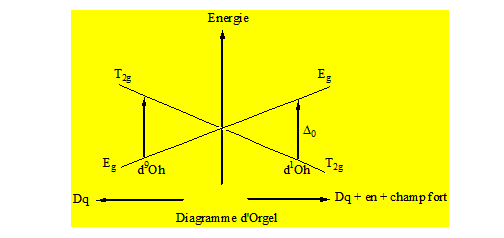
Soit le complexe [Cu(H2O)6]2+;
Cu2+ ; 3d9 → 2D
Transition Eg → T2g.
Si on associe à un métal donné des ligands de plus en plus champ fort on constate une différence de plus en plus grande entre T2g et Eg.
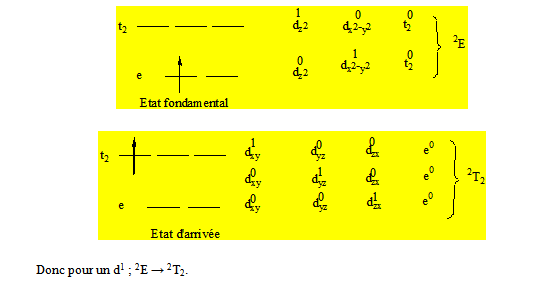
Si on considère Cu2+ qui est d9 on a un diagramme inverse par rapport à celui de Ti3+ qui est d1. Un tel diagramme représentant l’énergie en fonction des composantes est appelé diagramme d’Orgel. Maintenant pour le diagramme d’un d1 et d9 tétraédrique 2D → 2E + 2T2.
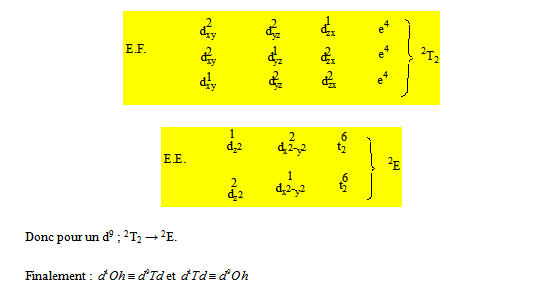
Pour un d9 on a :
Quand 2 métaux ont sur leur sous – couche d des nombres d’électrons complémentaires le diagramme d’Orgel de l’un en configuration tétraédrique est l’équivalent du diagramme d’Orgel de l’autre en configuration octaédrique ; alors que le diagramme d’Orgel de l’un en configuration tétraédrique ou octaédrique est l’inverse du diagramme d’Orgel de l’autre en configuration tétraédrique ou octaédrique. Les diagrammes d’Orgel permettent de rendre compte des spectres des complexes de type « high spin ». Pour les complexes de type « low spin » on fait intervenir les diagrammes de Tanabé – Sugano.
4 Diagramme de Tanabé - Sugano
Il n’est plus possible dans le cas des complexes de type « low spin » d’appliquer la règle de Laporte ou règle des électrons célibataires (les transitions entre les niveaux énergétiques provenant de la même sous – couche d sont interdites). Il faudra raisonner sur les termes de Russell – Saunders correspondant à l’état fondamental en considérant un complexe « low spin ».
Soit l’exemple de [Co(NH3)6]3+ qui est un complexe diamagnétique. Les termes de Russell – Saunders de Co3+ sont : 5D 3H 3F 3G 1I 1G 1S.
Si on considère le diagramme de Tanabé – Sugano d’un d6 on voit qu’il contient le diagramme d’Orgel d’un d6 « high spin ». C’est la première partie de ce diagramme. On voit en fait que le terme de Russell – Saunders à l’état fondamental dans le cas d’un « high spin » est 5D qui éclate en 5T2g et 5Eg. Ceci permet de rendre compte de la bande unique des spectres d’un complexe « high spin » d’un d6. Quand le complexe est « low spin » dans le cas d’un d6 le terme de Russell – Saunders à l’état fondamental n’est plus 5D mais 1I. Si on considère le remplissage des orbitales t2g dans un environnement Oh on voit qu’il n’y a qu’une seule façon d’écrire cet arrangement c’est – à – dire :
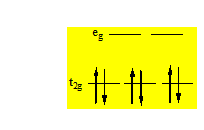
C’est pour cela que la composante fondamentale est A1g. Et l’ensemble des transitions partiront de cette composante fondamentale vers les autres composantes dérivant des termes de Russell – Saunders de multiplicité 1.
En réalité le spectre de [Co(NH3)6]3+ comporte 2 bandes. Si on considère une première transition simple on obtient les configurations :
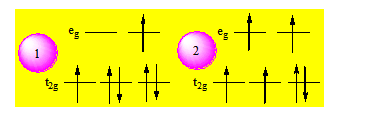
La configuration 1 est triplement dégénérée on l’appelle T1g. Si on considère un double saut électronique on obtient 2 qui est triplement dégénérée et qu’on appellera T2g. Les 2 absorptions présentes sur le spectre de [Co(NH3)6]3+ correspondant bien aux transitions : A1g → T1g ; A1g → T2g. Les autres transitions bien que possibles théoriquement ne peuvent pas être décelées par le spectrophotomètre U.V.
5 Couplages spin – orbite
Pour trouver les niveaux d'énergie issus de l’action du couplage spin - orbite sur un terme donné, il faut donc calculer toutes les valeurs possibles du nombre quantique MJ associé à et en déduire les valeurs possibles de J sachant que pour chaque valeur de J, MJ prend les 2J+1 valeurs J, J-1, . . .,-J + 1,-J. Le niveau obtenu est noté en ajoutant au terme dont il est issu l’indice J. Par exemple pour le terme fondamental 3F de la configuration d2, nous avons:
S = 1, d'où MS = ±1, 0
L = 3, d'où ML = ± 3, ± 2, ±1, 0
On en déduit que les valeurs possibles de J sont :
J = 4 auquel correspond MJ = ± 4, ± 3, ± 2, ±1, 0
J = 3 auquel correspond MJ = ± 3, ± 2, ±1, 0
J = 2 auquel correspond MJ = ± 2, ±1, 0
Les niveaux issus de 3F auxquels on donnera le nom d'état seront notés 3F4, 3F3 et 3F2. La dégénérescence 2J +1 de chacun de ces états ne peut être complètement levée que par l’action d'une perturbation externe, par exemple un champ magnétique.
Une règle empirique proposée à nouveau par Hund permet de prévoir l’état fondamental. Si la couche est moins que demi - remplie (configuration d1 à d4), l’état fondamental est donné par la plus petite valeur de J; si la couche est plus que demi - remplie, c’est la plus grande valeur de J qui correspond à l’état fondamental. Cet état fondamental, dans le cas de l’ion d2, est donc3F2.
Le couplage spin - orbite non seulement lève partiellement la dégénérescence de chaque terme mais peut aussi coupler des termes n’ayant ni les mêmes symétries orbitales et ni les mêmes multiplicités de spin en mélangeant les états ayant le même J. Cet effet du second ordre est d’autant plus important que le couplage spin - orbite est plus fort et que les termes sont moins espacés, c’est-à-dire que la répulsion électronique est faible. Ce couplage de différents termes est ainsi à peu près négligeable pour les ions de transition de la première série, il joue un certain rôle avec ceux de la seconde série et plus encore de la troisième.
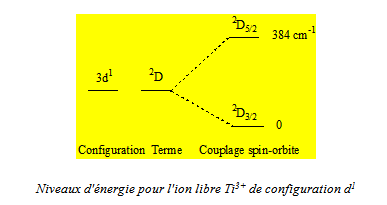
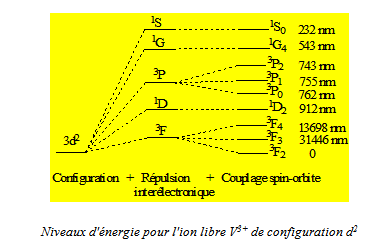
A titre d’exemple, nous représentons les effets successifs des différentes perturbations dans le cas d’une configuration d1, il n’intervient alors que le couplage spin - orbite, et d’une configuration d2. Bien que pour la clarté de la figure l’échelle des énergies n’ait pu être respectée, nous précisons les valeurs des énergies à partir de l’état fondamental obtenues expérimentalement pour les ions Ti3+ (d1) et V3+ (d2) et cela afin de bien mettre en évidence les importances relatives de la répulsion électronique et du couplage spin - orbite.
6 Propriétés optiques des composés de coordination (transitions d-d et interprétation qualitative des spectres)
La couleur des complexes résulte de transitions intraconfigurationnelles entre orbitales d. En effet, la différence d'énergie entre orbitales t2g et eg égale à Δ varie entre 12 500 cm-1 et 25 000 cm-1, c'est-à-dire une gamme d'énergie correspondant au spectre visible du rayonnement électromagnétique (800 nm > λ > 400 nm). Une transition électronique correspond au passage d'un électron du niveau fondamental vers un niveau excité. Lorsqu'elle a lieu, la matière absorbe un photon dont l'énergie correspond rigoureusement à la différence d'énergie entre le terme fondamental et un terme excité. Elle suit également des règles de sélection : l'une liée au moment orbital et l'autre lié au moment de spin. Une transition autorisée a lieu si les orbitales impliquées au cours de ce processus sont telles que Δl = ± 1 et ΔS = 0.
Les transitions d - d sont interdites selon la première règle car Δl = 0, mais elles sont observées grâce à des mécanismes de relaxation liés essentiellement à des couplages électrons-noyaux (rôle des vibrations). Par contre une transition d - d avec changement de spin est interdite selon les deux règles. Là encore, elle peut être observée grâce à des mécanismes de relaxation beaucoup moins efficaces liés aux couplages entre moments de spin et moments orbitaux des électrons. Par conséquent, ces transitions sont beaucoup moins intenses que les transitions d - d permises de spin. Nous ne parlerons que des transitions permises de spin dans la suite.
Le spectre électronique (mesure de la variation de l'absorbance d'une solution en fonction de λ) peut être formé d'une ou de plusieurs transitions électroniques.
Si un complexe ne présente qu'une seule transition, alors la couleur de ce complexe correspond à la couleur complémentaire de la couleur absorbée pendant la transition.
6-1 Spectres à une bande : interprétation de la couleur des complexes
Cette situation se produit dans les complexes des ions Ti3+ de configuration d1. Une solution aqueuse de [Ti(H2O)6]3+ est rouge. Le spectre d'absorption est formé d'une seule bande avec λmax = 510 nm. La couleur absorbée est le vert, le complexe apparaît de la couleur complémentaire à savoir le rouge. La bande observée est interprétée comme la transition de l'électron de l'orbitale t2g vers l'orbitale eg. L'énergie de cette transition est égale à Δ. On obtient donc la valeur de Δ à 510 nm, soit en unités cm-1, 19 607 cm-1.
Un cas similaire est celui de l'ion hexa-aquo de cuivre (II) : [Cu(H2O)6]2+ de configuration d9. Dans ce cas, le composé est bleu-vert, et son spectre est constitué d'une seule bande située à 840 nm. Il absorbe le rouge, et apparaît donc de la couleur complémentaire. Le spectre s'analyse de façon semblable. On peut voir cette transition comme le passage de l'électron d'une orbitale t2g à une orbitale eg. La transition possède toujours l'énergie Δ. Dans ce cas, Δ vaut 12 000 cm-1 (λ = 833,3 nm).
De l'analyse de ces deux spectres, on donne une preuve expérimentale que le champ cristallin Δ augmente avec la charge de l'ion de transition. De manière générale, ΔM3+ > ΔM2+.
6-2 Spectre à deux bandes : rôle de la répulsion « électron-électron »
Examinons maintenant le spectre visible du complexe [Cr(H2O)6]3+ dont la configuration de plus basse énergie est t2g3 (d3). Celui-ci est formé de deux bandes d'intensité semblable à 17 400 cm-1 (λ = 575 nm) et 24 600 cm-1 (λ = 406,5 nm), dont l'origine est à chercher dans des transitions t2g → eg. Une façon de réaliser cette transition est de promouvoir l'électron de l'orbitale dxy vers l'orbitale dz2. Dans ce cas, la densité électronique est relocalisée au cours de la transition d'un plan xy dans une direction z déjà riche en électron (les orbitales dzx et dyz sont occupées). Mais il existe une autre façon de réaliser cette transition en faisant passer l'électron de l'orbitale dzx vers l'orbitale dz2. Dans ce cas, la densité électronique sera simplement relocalisée selon un axe z déjà riche en électron avant la transition. On conçoit aisément que pour le premier cas la répulsion électronique va beaucoup augmenter au cours de la transition, et beaucoup moins dans le second cas. Par conséquent les deux transitions envisagées apparaissent à des énergies différentes.
Les autres possibilités de transitions t2g → eg seront similaires à l'un ou l'autre cas, si bien que le complexe possède deux bandes dans son spectre d'absorption.
En le premier cas, la densité électronique se déplace fortement vers les ligands sur l'axe z. Alors qu'au deuxième cas, le déplacement est beaucoup plus faible.
Une question vient à l'esprit immédiatement, quelle transition va permettre de déterminer Δ? Intuitivement, on choisira la transition de plus basse énergie, et on aura raison. On détermine donc Δ = 17 400 cm-1 (λ = 575 nm).
7 Interprétation quantitative des spectres
Pour comprendre l'origine de la deuxième transition pour le complexe [Cr(H2O)6]3+, il faut étudier plus en détail la structure électronique des ions de transition et tenir compte de la répulsion électron-électron. Jusqu'à présent celle-ci a été introduite dans le cas où elle est maximum, c'est-à-dire quand deux électrons occupent la même orbitale, sous la forme d'une quantité énergétique P. Lorsque P est beaucoup plus grand que Δ0, la structure électronique de l'ion considéré est profondément modifiée : le complexe à champ fort préfère associer tous ses électrons dans des orbitales t2g par souci de gain en énergie. Lorsque deux électrons occupent deux orbitales différentes orientées dans l'espace dans une direction commune, la répulsion électron-électron est plus faible mais elle existe. Elle aura pour effet de créer à partir d'un état dégénéré issu d'une configuration plusieurs niveaux d'énergies différentes.
Une configuration donnée (t2g)a(eg)b donne naissance, à cause de la répulsion « électron-électron », à plusieurs niveaux d'énergie différents, représentés par un terme spectroscopique. Les transitions entre termes spectroscopiques ont des énergies couvrant le domaine du visible. Les spectres optiques des composés de coordination ne s'interprètent donc pas comme un saut électronique t2g → eg, mais comme une transition entre termes spectroscopiques (T2g → Eg).
De la même manière que nous avons comparé P et Δ0 (= 10 Dq) jusqu'à présent, nous allons comparer la répulsion électron-électron traitée dans sa globalité (qui sera notée par la suite RE) et le champ cristallin. Commençons par examiner deux cas limite : la situation champ faible Δ0 << RE et la situation champ fort Δ0 >> RE.
7-1 Structure électronique des complexes à champ faible
Le champ cristallin est faible, les effets de la RE dominent : Δ0 << RE. Nous considérons d'abord l'effet de la RE sur les niveaux d'énergie de l'ion libre. Les cinq orbitales d (ou si l'on tient compte du spin les dix spin-orbitales) sont toutes de même énergie. Pour la configuration d3, il faut placer trois électrons (ou trois objets) parmi dix spin-orbitales (ou dix cases). La statistique permet d'évaluer le nombre de possibilités de ranger p objets dans n cases, selon :
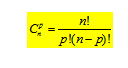
Avec n, nombre de spin-orbitales et p, le nombre d'électrons. Pour d3, n = 10, p = 3, soit . Il existe 120 possibilités de ranger 3 électrons sur 10 spin-orbitales.
La configuration d3 regroupe à elle seule 120 états d'énergie équivalente égale à trois fois l'énergie d'une orbitale d, soit 3.Ed. La RE va lever cette dégénérescence en créant huit niveaux d'énergie différents qui sont les termes spectroscopiques.
7-2 Structure électronique des complexes à champ fort
Δ0 >> RE, le champ cristallin est fort, et ses effets sur les niveaux d'énergie dominent.
L'effet du champ cristallin sur une configuration dn est traité dans un premier temps. Ainsi les orbitales d deviennent en champ octaédrique les orbitales t2g d'énergie -2/5 Δ0 et les orbitales eg d'énergie -3/5 Δ0. A partir de la configuration d3, on obtient les configurations d'énergie et de dégénérescence différente :
- (t2g)3 d'énergie égale à 3(-2/5 Δ0) = -6/5 Δ0,
- (t2g)2(eg)1 d'énergie égale à 2(-2/5)Δ0 + 3/5Δ0 = - 1/5 Δ0,
- (t2g)1(eg)2 d'énergie égale à -2/5Δ0 + 2(3/5Δ0) = 4/5 Δ0,
- (eg)3 d'énergie égale à 3(3/5Δ0) = 9/5 Δ0.
La RE a pour effet de lever partiellement cette dégénérescence pour créer les séries de termes spectroscopiques déjà identifiés dans la première situation. Cette situation donne accès à l'énergie des termes.
7-3 Corrélation « champ faible-champ fort ». Diagrammes de Tanabé-Sugano
La réalité étant très souvent intermédiaire entre un champ faible et un champ fort, on établit une corrélation termes à termes de l'approche champ faible vers l'approche champ fort (figure suivante).
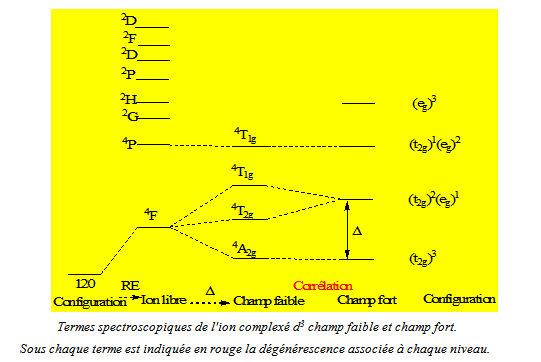
Cette corrélation aboutit à la construction d'un diagramme de Tanabé-Sugano (figure ci - après), où l'énergie des termes est portée selon E/B = f (Δ/B), B (terme relatif à la répulsion électronique) étant le paramètre de Racah. Le terme fondamental est ainsi confondu avec l'axe des abscisses.
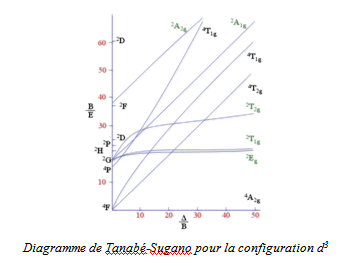
7-4 Interprétation
Interpréter un spectre signifie que l'on est capable d'identifier les termes excités atteints lors de chacune des transitions en considérant le diagramme de Tanabé - Sugano décrivant la configuration étudiée. Une interprétation qualitative du spectre de [Cr(H2O)6]3+; (configuration d3, diagramme figure précédente) est la suivante :
Le terme fondamental est 4A2g, terme issu de la configuration de plus basse énergie (t2g)3.
La configuration excitée de plus basse énergie est (t2g)2(eg)1. La RE va créer deux termes spectroscopiques de même multiplicité de spin que le terme fondamental 4T2g et 4T1g. On obtient deux transitions dans le domaine du visible :
- 1ère transition : 4A2g → 4T2g ;
- 2ème transition : 4A2g → 4T1g ;
A priori, une troisième transition devrait être observée, correspondant à la transition entre l'état fondamental 4A2g et le terme excité 4T1g le plus haut en énergie. Dans les faits, cette transition est trop haute en énergie pour être observée dans le domaine du visible.
On remarque que l'on a volontairement négligé la transition vers les termes excités 2Eg, 2T1g. En effet la transition du terme fondamental 4A2g vers ces deux termes de même énergie est une transition qui modifie le spin du système. Ce type de transitions est interdit selon les règles de sélection, donc à priori non observable. Or, si on examine le spectre de manière attentive, il existe un épaulement dans le pied de la bande correspondant à la transition 4A2g → 4T2g ; situé en dessous de 16 000 cm-1 (λ = 625 nm).
Cet épaulement correspond à la transition 4A2g → 4Eg, 2T1g peu intense masquée par la transition permise de spin. Des mécanismes de relaxation mentionnés au début de ce chapitre sont à l'origine de son observation.
Une fois l'analyse qualitative effectuée, l'interprétation quantitative du spectre permet de déterminer Δ et B. Pour cela, nous allons effectuer une résolution graphique.
Pour déterminer les deux inconnues Δ et B, nous allons effectuer une résolution graphique. Nous remarquons sur le diagramme que la pente du premier terme excité 4T2g est égale à un. Par conséquent, on a pour ce terme E2/B = 1. Δ/B, soit E2 = Δ. La première transition nous donne directement la valeur de Δ, soit Δ= 17400 cm-1. Pour estimer B, nous calculons le rapport (E2/B)/(E1/B) = E2/E1, soit 24600/17400 = 1,41. Cette astuce nous permet d'éliminer l'inconnue B sur un des axes. On cherche alors graphiquement le rapport correspondant E2/E1 = 1,41 sur l'axe des ordonnées, et quand il est trouvé, il suffit de lire sur l'axe des abscisses la valeur de Δ/B, soit dans ce cas 25. On trouve donc pour B = 700 cm-1.
8 Propriétés magnétiques des composés de coordination
8-1 Description macroscopique et microscopique du magnétisme de composés moléculaires
Une autre façon d'appréhender la force du champ cristallin est de mesurer les propriétés magnétiques des complexes. Deux types de comportements magnétiques sont attendus pour les complexes : le paramagnétisme (le composé est attiré vers la zone de champ magnétique maximum) et le diamagnétisme (le composé est repoussé vers la zone de champ magnétique minimum). Il s'agit de magnétisme induit, c'est-à-dire que la présence d'un champ magnétique extérieur est nécessaire pour mettre en évidence ces comportements. Il existe deux types de comportements magnétiques pour deux complexes de l'ion Fe2+ (configuration d6) : le complexe [Fe(H2O)6]2+ présent dans FeSO4.7H2O et le complexe K4[Fe(CN)6]. Dans le premier cas, il existe des électrons non appariés, le complexe est donc spin fort-champ faible. Dans le deuxième cas, les électrons sont tous appariés, le complexe est donc « spin faible-champ fort ».
8-2 Mesures magnétiques et loi de Curie
Toute molécule résulte d’une association d’atomes, eux-mêmes constitués chacun d’un noyau chargé positivement autour duquel gravitent des électrons chargés négativement. Il existe une étroite relation entre la structure électronique et les propriétés magnétiques d’une molécule.
L’étude des propriétés magnétiques permet de déterminer la structure des complexes des ions métalliques ainsi que la nature (anti ou ferromagnétique) de l’interaction entre deux ou plusieurs centres métalliques. Ainsi, il existe une étroite relation entre la structure et les propriétés magnétiques d’une molécule. Le magnétisme moléculaire est une technique qui permet d’appréhender la géométrie et la structure électronique des ions métalliques à partir de la susceptibilité magnétique.
Pour étudier les propriétés magnétiques d’une substance, on la soumet à l’action d’un champ magnétique intense. L’échantillon acquiert alors une aimantation. En effet en présence d’un champ magnétique , les différents moments magnétiques électroniques ou nucléaires vont se diviser en différents niveaux d’énergie. Pour le noyau d’hydrogène caractérisé par un spin de valeur 1/2, l’aimantation peut prendre deux positions dites parallèle ou anti parallèle. L’état parallèle étant de plus basse énergie, il est plus peuplé et il en résulte dans le milieu, une aimantation nucléaire macroscopique notée M (moment magnétique par unité de volume). Cette aimantation est proportionnelle à l’intensité du champ magnétique appliqué. Le coefficient de proportionnalité, noté c, définit la susceptibilité magnétique du milieu considéré : M = cH. Lorsque c est positif on dit que le corps dans lequel apparaît l’aimantation est paramagnétique; cet effet provient des électrons non appariés de la substance. Lorsqu’ il est négatif, le corps est dit diamagnétique ; la substance est légèrement repoussée par le champ magnétique, cet effet provient des électrons appariés. Le magnétisme moléculaire est donc une technique qui permet de déterminer l’état de perturbation d’un ion métallique à partir de la susceptibilité.
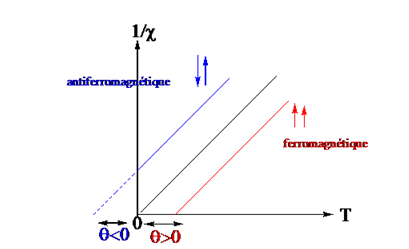
La susceptibilité diamagnétique, présente dans tous les composés, est négative et indépendante de la température. Elle est liée à la circulation des électrons appariés dans leurs orbitales induite par le champ magnétique externe. Elle se calcule à partir de contributions atomiques constituant la substance étudiée. La susceptibilité paramagnétique existe uniquement pour les composés possédant des électrons non appariés. Elle est positive et dépendante de la température. Elle est beaucoup plus importante que la susceptibilité diamagnétique.
Pour les complexes, il est utile de définir une susceptibilité molaire notée χM.
Les susceptibilités diamagnétiques molaires par atome ou groupe d’atomes sont données au tableau suivant :
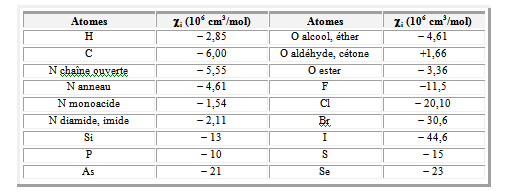
La dépendance thermique de la susceptibilité paramagnétique molaire est donnée par la loi de Curie, selon la relation :
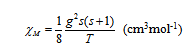

Par exemple, le complexe [FeII(H2O)6]2+ possède un spin s = 2 et g = 2, donc le produit χMT vaut 3 cm3mol-1K. Le complexe diamagnétique [FeII(CN)6]4- possède un spin s = 0, d'où χMT est nul. Le complexe [CrIII(H2O)6]3+ a trois électrons célibataires dans les orbitales t2g, donc s = 3/2, soit χMT = 1,875 cm3mol-1K avec g = 2. Ainsi, la valeur du produit χMT permet de connaître le nombre d'électrons célibataires dans un composé.