Imprimer tout le livre
Imprimer tout le livre
Chimie Organique I
description du cours
| Site: |
Touch By SukaJanda01 |
| Cours: |
Chimie Organique (Année 2015) |
| Livre: |
Chimie Organique I |
| Imprimé par: |
Visiteur anonyme |
| Date: |
lundi 16 juin 2025, 11:04 |
1 Généralités
Durée: 1semaine
Objectifs: comprendre l'histoire de la chimie et définir la chimie organique.
1.1 Introduction
En tant qu’outil important pour nous permettre d’accéder à la connaissance du monde qui nous entoure, la chimie — l’étude de la matière et des règles qui régissent les changements qu'elle effectue — s’est développée lentement jusque vers la fin du XVIIIe siècle. À cette époque, en se basant sur ses études du phénomène de la combustion, Antoine-Laurent de Lavoisier apporta des indices que les compositions chimiques
pouvaient être déterminées par l’identification et la mesure des quantités d’eau, de dioxyde de carbone et d’autres matières produites lorsque les substances sont brûlées dans l’air. À cette époque, la science de la chimie comportait déjà deux grandes divisions: l’une traitant des substances obtenues des sources naturelles ou vivantes, et l’autre traitant des substances dérivées de la matière non vivante. Il s’agissait respectivement
de la chimie organique et de la chimie inorganique. Les études de la combustion ont rapidement établies que les composés dérivés de sources naturelles comportaient toujours du carbone. Ceci a amené une nouvelle définition de la chimie organique: l’étude des composés du carbone. Cette définition est encore utilisée aujourd’hui.
Les domaines actuels de la chimie sont les suivants: chimie analytique, biochimie, chimie inorganique, nucléaire, organique, physique, chimie de polymères, chimie théorique.
Les molécules organiques constituent l’essence même de la vie. Les protéines, les acides nucléiques, les sucres et les graisses sont des composés dont le constituant principal est le carbone. Les vêtements que nous portons sont faits de polymères naturels (coton, soie) ou synthétiques (polyesters). Les produits domestiques d’usage courants, dentifrices, savons, shampooings, déodorants, parfums sont tous des produits de l’industrie chimique organique. On peut y inclure aussi les peintures, plastiques, aliments, etc. Il ne faut pas oublier aussi toute la panoplie de produits pharmaceutiques (médicaments, vaccins, antibiotiques) et autres insecticides et pesticides.
1.2 Définitons
•La chimie organique est la branche de la chimie qui concerne la description et l’étude des composés organiques.
•Attention: ne pas confondre avec la biochimie, qui étudie les molécules fabriquées par les organismes vivants.
•Les composés organiques sont des molécules composées d’atomes de carbone et d’hydrogène.
•En plus des atomes de carbone et d’hydrogène, les molécules organiques peuvent contenir des atomes d’oxygène, d’azote, de soufre, des halogènes (chlore, fluor, brome,…) ainsi que plus rarement autres atomes.
•Les atomes qui composent ces molécules sont toujours liés entre eux par des liaisons covalentes: elles peuvent être simples, doubles voire triples!
Exemple de molécule organique:
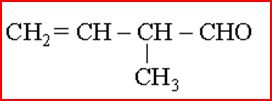
1.3 Quelques vocabulaires
•Saturation: une molécule saturée n’est composée que par des liaisons simples, alors qu’une molécule insaturée possède au moins une liaison double.
•Conjugaison: un composé conjugué est un composé où se produisent des déplacements d’électrons le long des liaisons covalentes. Exemple: une molécule où deux doubles liaisons sont séparées par une simple liaison:

•Polarité: une liaison covalente polarisée voit ses électrons se rapprocher d’un des atomes sous l’influence de plusieurs facteurs (électronégativité, effets inductif ou mésomère). Ceci a un grand impact sur la réactivité de la liaison.
•Aliphatique: une chaîne aliphatique ne contient aucune conjugaison (linéaire ou cyclique).
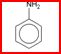
•Aromatique: une molécule aromatique contient un cycle d'atomes de carbone similaire à celui du benzène (doubles liaisons conjuguées). Exemple: aniline
2 Nomenclature
Durée: 2 semaines
Objectif: structurer un composé organique connaissant son nom.
2.1 Nomenclature des hydrocarbures saturés
•Pour pouvoir nommer un composé organique, il faut suivre des règles précises, qui sont à la base de la chimie organique.
•D’abord, il faut trouver la chaîne hydrocarbonée la plus longue à considérer comme l'alcane de base et lui donner un nom. Les plus communs: 1C=méthane, 2C=éthane, 3C= propane, 4C=butane. Après, les préfixes habituels: pent-, hex-, hept-, oct-, non-,…Si la molécule forme un cycle, faire suivre l’alcane du préfixe cyclo-.
•Ensuite il faut identifier tout groupe alkyl (ex: éthyl CH3-CH2-) branché sur l'alcane et lui donner un nom (radical); placer ce nom en préfixe devant le nom de l'alcane de base.
•Finalement, faire suivre le nom de chaque radical par un chiffre indiquant la position de la ramification sur l'alcane de base (s’il y a plusieurs ramifications, alors il faut prendre la somme des indices la plus faible).
Exemple: éthyl-4 méthyl-3 heptane: 
2.2 Nomenclature des hydrocarbures insaturés
•Suit les mêmes règles que pour un hydrocarbure insaturé, il suffit de changer le suffixe.
•Pour un alcène simple (double liaison C=C) le nom de la molécule termine par –ène, suivi par le chiffre indiquant la position de l’insaturation. S’il y a deux doubles liaisons, alors le suffixe est –diène. Exemple: butadiène-1,3 CH2=CH-CH=CH2
Les molécules cycliques aromatiques ont une nomenclature particulière dont la plus connue est celle du benzène

2.3 Nomenclature des composés contenant au moins un groupement fonctionnel
•Lorsque le composé contient un seul groupement fonctionnel, alors il est généralement signalé en suffixe (suivi de sa position si nécessaire).
•Si le composé contient deux ou plus groupements fonctionnels, alors seule la fonction principale est indiquée en suffixe, les autres apparaissant en tant que préfixes (une hiérarchie existe entre les fonctions).
•Exemple des alcools: si principale alors suffixe –ol, si secondaire alors préfixe hydroxy- (suivies si nécessaire du numéro indiquant leur position).
•Certaines fonctions sont toujours indiquées en préfixe. Exemple: les dérivés halogénés (fluoro-, chloro-, bromo-, iodo-).
3 Principales fonctions organiques.
Durée: 2 semaines
objectif: Classifier les fonctions organiques.
3.1 Alcools
2.4.1. Alcools
•Formule générale: R-OH
•Trois classes:
1) Alcool primaire: R-CH2-OH (OH en bout de chaîne)
2).Alcool secondaire: R-CHOH-R’
3).Alcool tertiaire:

•Liaison polarisée en faveur de l’oxygène (effet inductif attracteur).
•
Nomenclature: suffixe –ol ou préfixe hydroxy- (suivis du chiffre indiquant la position si nécessaire).
•Acidité très faible: départ du H très difficile, ne réagit pas avec la soude.
•Subissent des réactions d’oxydation:
Alcool primaire → aldéhyde → acide carboxylique
Alcool secondaire → cétone
3.2 Thiols
•Composés soufrés avec des propriétés très similaires à celles des alcools (en effet, O et S sont situés sur la même colonne du tableau périodique).
•Liaison polarisée en faveur du soufre (effet inductif attracteur).
•Formule générale: R-SH
Oxydation: modérée, conduit à la formation de thioéthers R-S-S-R (rôle dans les protéines au niveau des ponts disulfure).
3.3 Aldéhydes – cétones
•Composés carbonylés: double liaison avec un carbone.
•Aldéhydes: oxygène en bout de chaîne R-CH=O
•Cétones: oxygène en milieu de chaîne:

•Liaison polarisée en faveur de l’oxygène (effet mésomère attracteur).
•
Nomenclature: suffixe –al pour un aldéhyde (pas de numéro car toujours en bout de chaîne) et –one pour une cétone (suivie du chiffre de sa position si nécessaire).
•Réduction: d’un aldéhyde en alcool primaire, d’une cétone en alcool secondaire.
•Oxydation: d’un aldéhyde en acide carboxylique. Une cétone ne peut s’oxyder que s’il y a rupture de la chaîne.
Exemples: butanone CH3-CO-CH2-CH3, propanal CH3-CH2-CHO
3.4 Acides carboxyliques
•Composés carboxylés: un carbone lié à deux atomes d’oxygène (une simple liaison et une double liaison).
•Formule générale: R-COOH:

•
Nomenclature: acide au début du nom suivi du suffixe –oïque.
•Acidité: en solution dans l’eau, l’acide se dissocie partiellement pour donner des ions carboxylate R-COO- (départ d’un proton H+).
•Mésomérie:

Dérivés: nombreux, par exemple les esters et les amides

3.5 Amines
•Composés azotés: simple liaison avec un atome d’azote.
•Formule générale: R-NH2
•Trois classes (attention: différent aux alcools):
1) Amine primaire: R-NH2
2) Amine secondaire: R1-NH-R2
3) Amine tertiaire:

•Liaison polarisée en faveur de l’azote (effet inductif attracteur).
•Basicité: dans l’eau les amines sont des bases faibles (captent un proton H+).
Autres composés azotés: les imines (double liaison avec l’azote), les nitriles (triple liaison), les amides (cf. ci-dessus).
3.6 Dérivés halogénés
•Halogènes: atomes de la septième colonne de la classification périodique (fluor, chlore, brome, iode).
•Formule générale: R-X
•Liaison polarisée en faveur de l’halogène (effet inductif attracteur).
Nomenclature: toujours en préfixe (l’halogène est considéré comme un substituant) halogéno- ou halogénure de. Exemple: dichlorométhane CH2Cl2.
4 Isomérie
Durée: 2 semaines
Objectifs:: Permettre à l'étudiant d'expliquer les différences structurales des composés de même formule brute aussi bien dans le plan que dans l'espace.
4.1 Les environnements des atomes en chimie organique
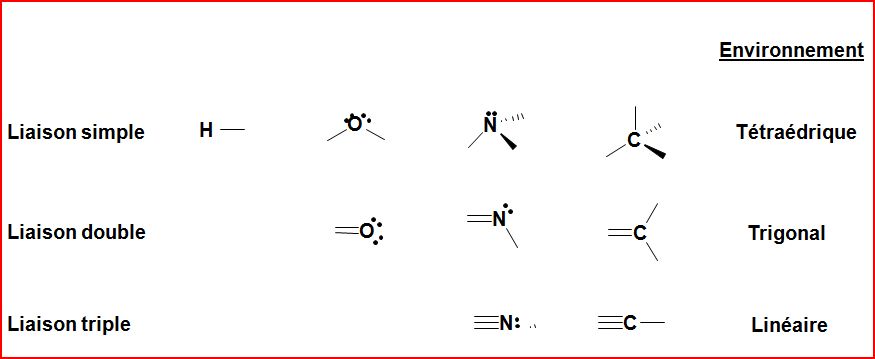
4.2 Formules de constitution
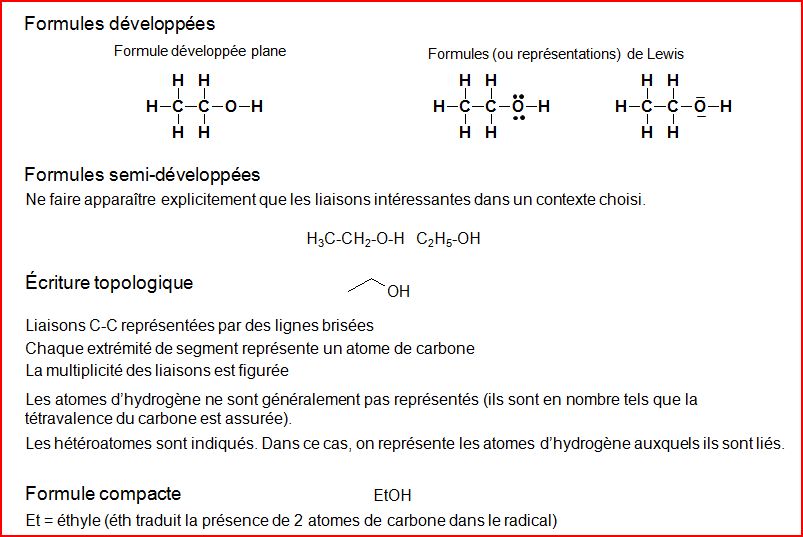
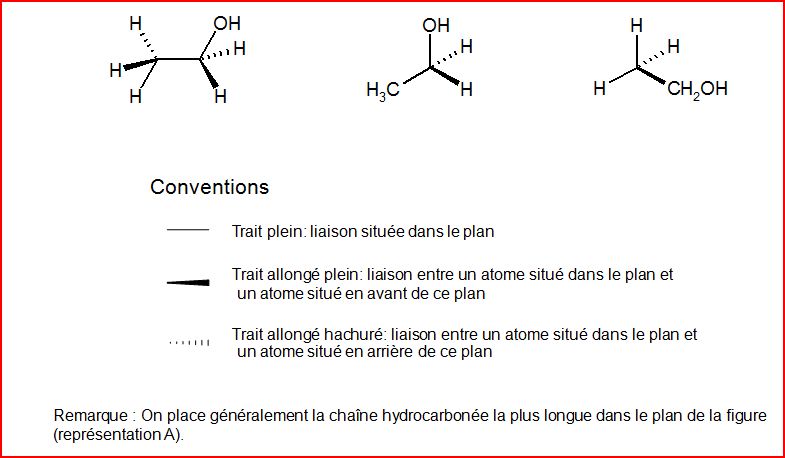
4.3 Représentation de CRAM
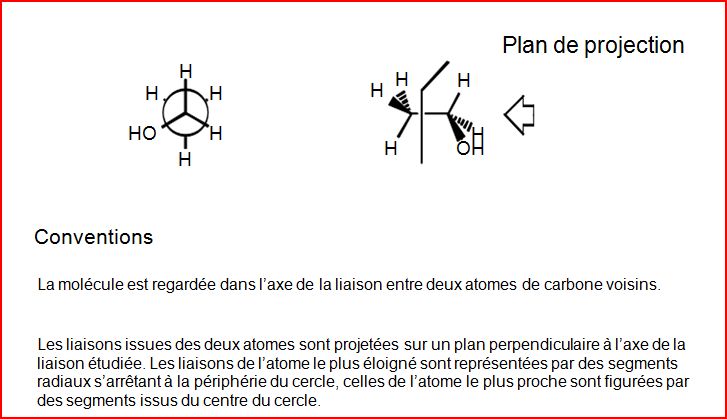
4.4 Représentation en projection de Newman
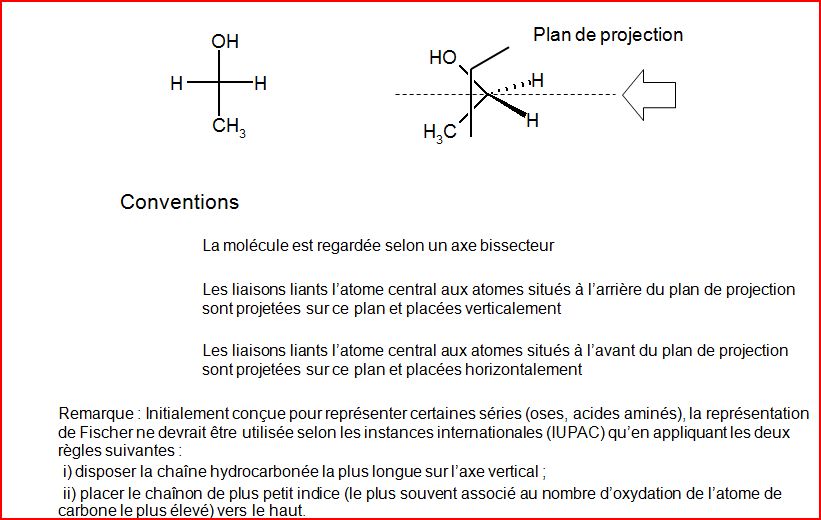
4.5 Représentation de Fischer
4.6 Notion d’isomérie
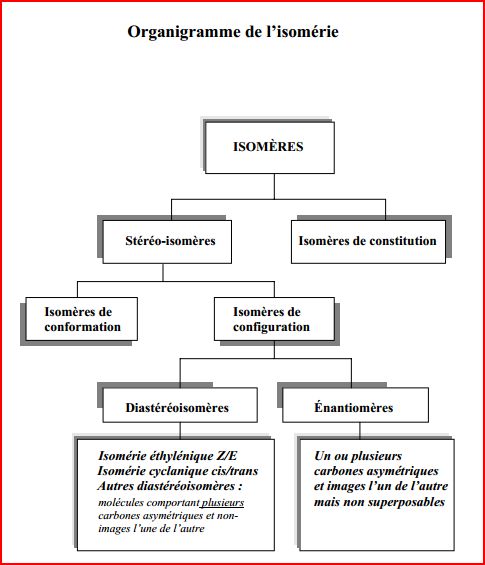
Les isomères sont des espèces chimiques de même formule brute qui diffèrent par :
- l’ordre ou la nature des liaisons (isoméries de constitution),
- ou par la disposition des atomes dans l’espace (stéréoisomérie).
4.7 Isomérie de constitution

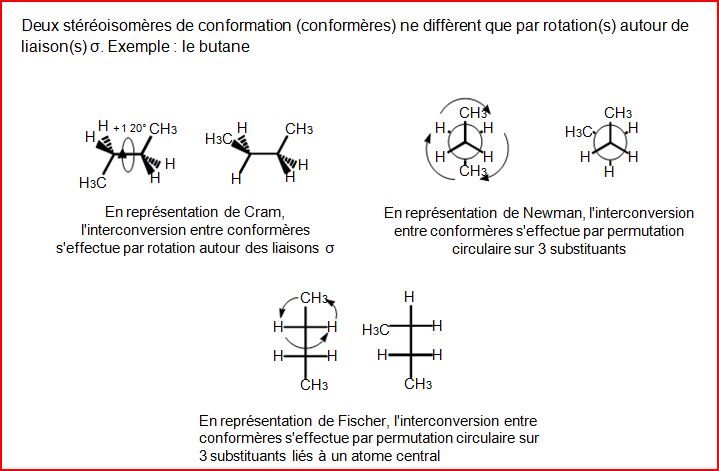
4.8 Stéréoisomérie de conformation
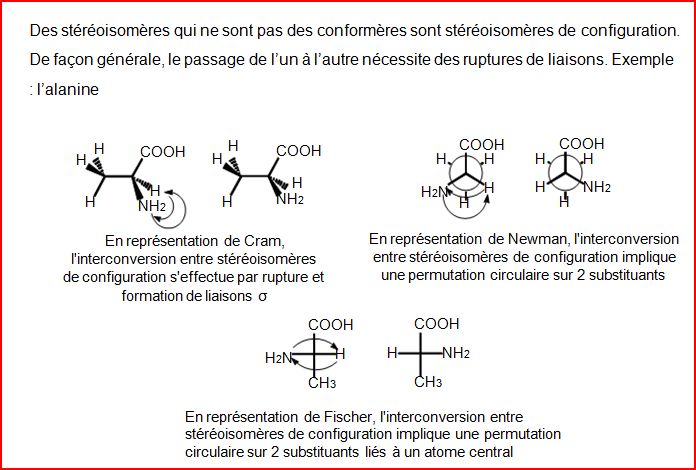
4.9 Stéréoisomère de configuration
4.10 Organigramme de l'isomérie
4.11 Nommer les stéréoisomères

4.10.1 Configuration Z ou E
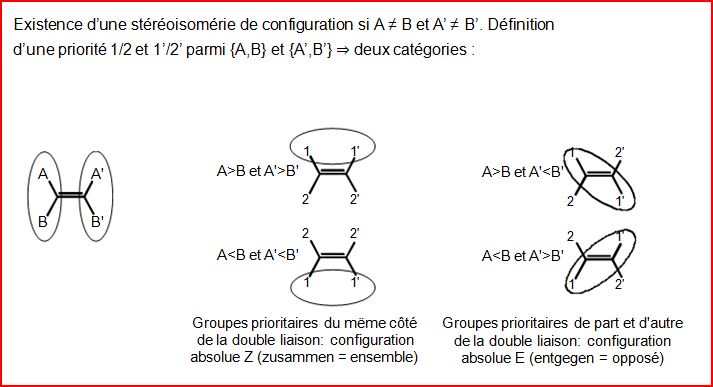
4.10.2. Configuration R ou S
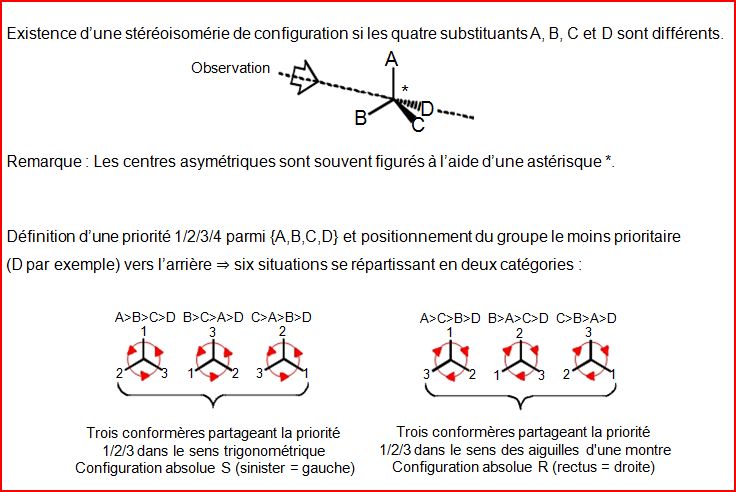
4.10.3. Règles de priorité Cahn-Ingold-Prelog

4.10.4 Exemples de détermination de configuration
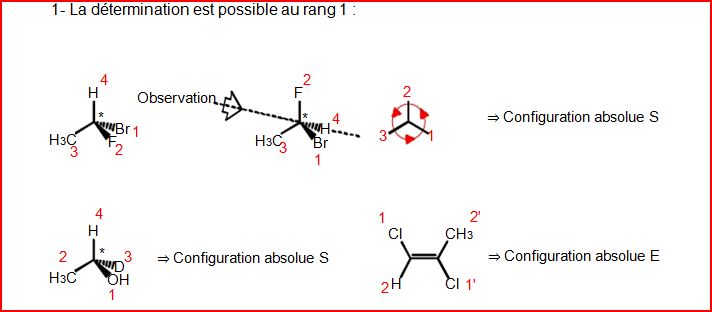
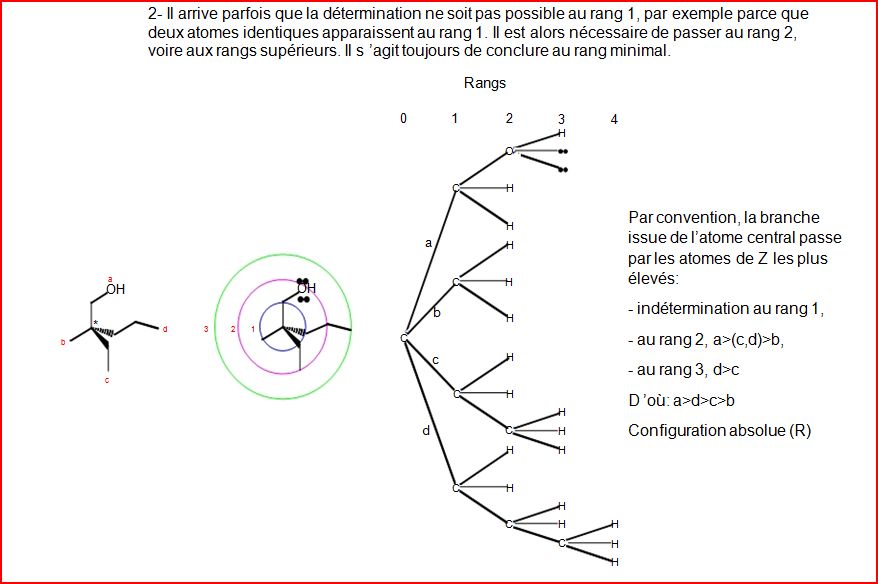
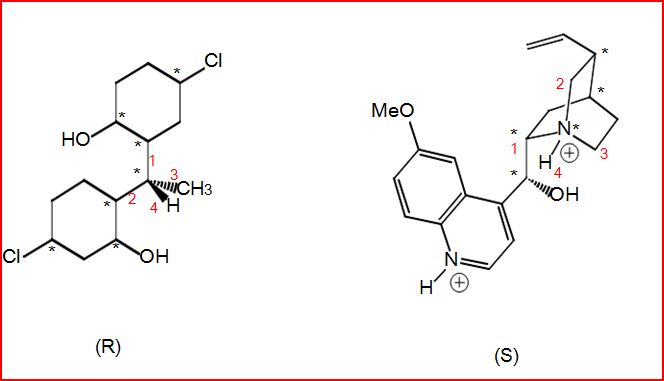
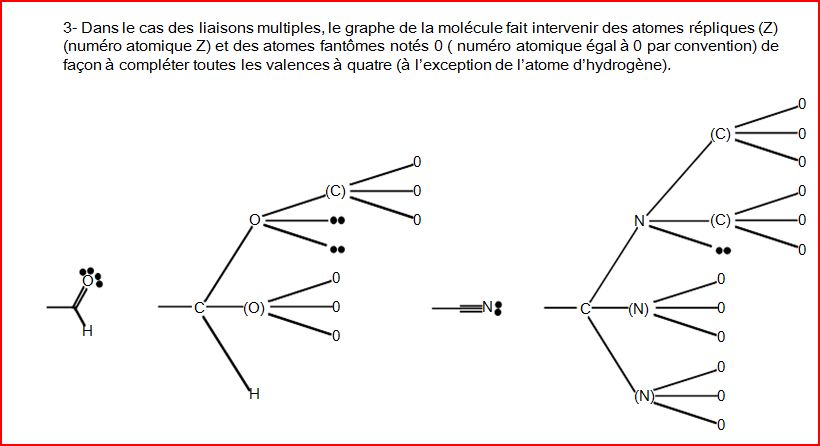
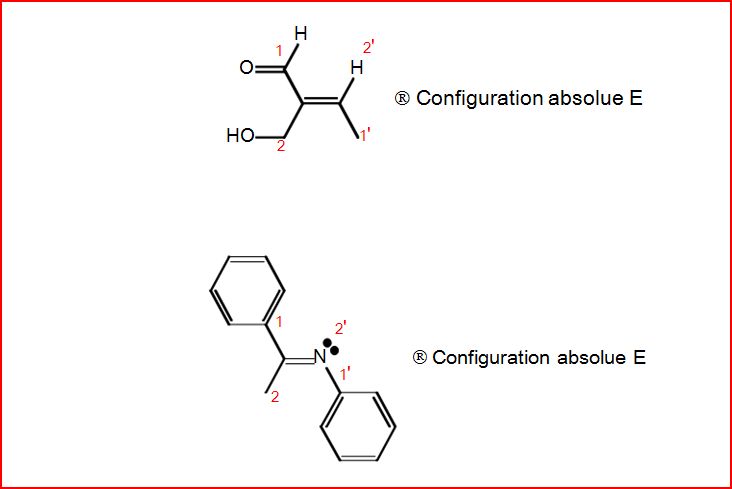
4.12 Chiralité
Le premier critère mis en place pour distinguer des catégories de stéréoisomères repose sur la nature des déformations appliquées pour passer d’un stéréoisomère à un autre : rotation autour d’une liaison/rupture d’une liaison.
Il existe un second critère qui repose sur des considérations de symétrie.
La chiralité d’un objet désigne sa propriété de ne pas être superposable à son image dans un miroir plan.
Un objet possédant un plan ou un centre de symétrie est achiral (non doué de chiralité).
Exemples : Une main est un objet chiral.
Une molécule contenant un carbone asymétrique est chirale
4.13.1. Molécule contenant un carbone assymétrique

4.13.2. Molécule contenant plusieurs carbones asymétriques

4.13.3. Enantiomérie

4.13.4. Diastéréoisomérie

4.13.5. Relation d'énantiomérie-diastéréoisomérie

4.13.6. Cas d'un composé méso

4.13.7. Activité optique

Deux énantiomères possèdent des pouvoirs rotatoires spécifiques opposés.
Un mélange racémique, c’est-à-dire un mélange équimolaire d’énantiomères, est donc inactif par compensation.
5 Effets électroniques
Durée: 2 semaines
Objectif: Interpréter les mouvements électroniques dans les molécules organiques.
5.1 Electronégativité
L’électronégativité augmente de la gauche vers la droite sur une même période, et du bas vers le haut sur une même colonne.

5.2 Sites électrophiles/nucléophiles
•Un site électrophile (symbolisé E+) est pauvre en électrons, il va donc essayer de « gagner » des électrons provenant d’un composé nucléophile (noté Nu-).
•Un site nucléophile possède une densité électronique élevée, il est donc susceptible de réagir avec un composé électrophile avide d’électrons.
•Toute liaison polarisée contient un site nucléophile correspondant à l’atome le plus fortement électronégatif (charge partielle δ-) et un site électrophile (charge partielle δ+).
Exemple: au niveau d’une liaison alcool, l’oxygène très électronégatif est le site nucléophile, alors que le carbone moins électronégatif constitue le site électrophile.
•En chimie organique, les réactions portent souvent un nom selon la nature du réactif qui attaque le substrat.
Exemple: dans une addition électrophile, un alcène (substrat) subit d’abord une attaque par un réactif électrophile (H+, Cl+, Br+,…).
5.3 Effet inductif
•Concerne uniquement les électrons σ et traduit l’action de déformation du nuage électronique σ le long d’une liaison simple. Il peut être attracteur (I-) ou donneur (I+)
•Il augmente avec la différence d’électronégativité entre les deux atomes. Ex: l’effet inductif attracteur du fluor dans F-CH3 est supérieur à celui du chlore dans Cl-CH3.
•Il se propage le long d’une chaîne en s’atténuant (disparaît au bout de trois liaisons covalentes).
•Il est cumulatif et est représenté par une flèche au niveau de la liaison indiquant le sens du déplacement des charges.
•Exemples de groupements exerçant un effet inductif:
Donneurs: CH3-, CH2-CH3-, Li-, Na-,…
Attracteurs: Cl-, OH-, Br-,…
•Exemples: H3C-CH=CH2 ; F-CH2-CH2-CH3
5.4 Effet mésomère
•Concerne les électrons

et les électrons non liants (p). L’effet mésomère peut être donneur (M+) ou attracteur (M-).
•Il traduit l’action de déformation du nuage électronique � dans un système conjugué permettant une délocalisation électronique.
•Il est plus intense qu’un effet inductif.
•Il se propage le long des liaisons conjuguées sans atténuation.
•Le phénomène de mésomérie (résonance) correspond à une stabilisation du système.
•Exemple de groupements impliqués dans un effet mésomère:
Donneurs: halogènes (par un de leurs doublets non liants), oxygène d’un alcool (idem), azote d’une amine (idem)…
Attracteurs: oxygène d’un aldéhyde ou d’une cétone, azote d’une imine… 6 Principaux mécanismes réactionnels
Durée: 2 semaines
Objectifs: Classer les réactions organiques et expliquer les types de ruptures de liaisons.
6.1 Principaux types de réactions organiques
Toutes les réactions en chimie organique sont le résultat d’un mécanisme simple ou de la combinaison de mécanismes simples.
. Addition: s’effectue sur une insaturation. Un réactif et son substrat se combinent pour former un seul produit (A+B→A-B)
Exemple: CH2=CH2 + Cl2 → CH2Cl-CH2Cl
. Élimination: un réactif donne deux produits, avec la formation d’une insaturation (A-B →A+B), souvent sous l’influence d’une température élevée.
Exemple: CH3-CH2-OH → CH2=CH2 + H2O
. Substitution: un réactif modifie et se lie sur son substrat pour obtenir une nouvelle molécule (A-B + C → A-C + B).
Exemple: CH3-CH2-OH + Cl- → CH3-CH2-Cl + OH-
• Exemple de combinaison de plusieurs mécanismes: estérification (cf. lycée): addition suivie d’une élimination.
6.2 Rupture des liaisons simples et intermédiaires réactionnels
•Selon la polarisation de la liaison covalente simple (σ), plusieurs cas de figure sont possibles.
•Liaison non polarisée: atomes de même électronégativité, par exemple dans les alcanes. Il se produit une rupture homolytique: chaque atome prend un électron célibataire du doublet liant, ce qui produit des radicaux libres. L’effet des UV est souvent nécessaire.
R-R → R. + R. (R.représente un radical libre)
•Liaison polarisée: atomes d’électronégativité différente. Il se produit une rupture hétérolytique: l’atome le plus électronégatif garde les électrons du doublet liant (exemple: dérivés halogénés). Il se produit une ionisation.
R-Br → R+ + Br-
7 Introduction à la Résonance Magnétique Nucléaire
Durée: 2 semaines
Objectif: Interpréter les spectres RMN 13C et 1H des composés organiques.
7.1 Définition
•Résonance Magnétique Nucléaire
•Permet la détection des Noyaux atomiques.
•Fournit des indications sur l’environnement à l’intérieur de la molécule
7.2 La RMN, Analogie avec la boussole
¨Les noyaux (1H, 13C) se comportent comme des petites boussoles
¨Ils ont 2 niveaux d’énergie: sur (faible énergie) ou contre (haute énergie) le champ
¨Une onde radio permet le passage entre ces 2 niveaux: Fréquence de résonance.
7.3 Fonctionnement de la RMN
•L’échantillon est dissous dans un solvant convenable et placé dans un champ magnétique intense
–Les protons ont maintenant 2 niveaux d’énergie: sur et contre le champ
•L’échantillon est irradié par une courte impulsion de radiofréquence
–Cela dérange l’équilibre entre les 2 niveaux d’énergie: certains absorbent de l’énergie et passent au niveau supérieur
•Détection de l’énergie libérée par le retour des noyaux à l’état inférieur
–Utilisation d’un récepteur radio sophistiqué
•Après calcul, les résultats sont affichés sous forme d’un graphe d’intensité en fonction de la fréquence.
Exemple de spectres RMN 13C et 1H du propan-1-ol
•Solvant: CDCl3, Aimant: 90 MHz
¨Les fréquences sont exprimées en ppm (partie par million) par rapport à la fréquence fondamentale de l’aimant. Ici 1 ppm = 90 Hz
L’échelle est orientée vers la gauche
7.4 Pourquoi les noyaux sont-ils différents?
•Signal = Fréquence de l’énergie absorbée par les noyaux qui dépend:
–Du type de noyau observé: 13C ou 1H
–De la force du champ magnétique: 90MHz, 400MHz, …
•Pour un même type, les atomes devraient absorber la même fréquence!!
•Mais NON!!! Car le champ magnétique subit par le noyau n’est tout à fait celui qui est appliqué
Le coupable = L’électron !!
7.5 Le blindage des noyaux par les électrons?
La répartition des électrons autour d’un noyau affecte
- Le champ magnétique local subit par le noyau, plus la densité électronique est forte moins le champ est fort (blindage)
- La fréquence à laquelle le noyau résonne, moins le champ est fort plus petite est la fréquence
Cette variation de la fréquence est appelée déplacement chimique (noté  )
)
7.6 Les déplacements chimiques: cas du propan-1-ol
- H proche de O implique (Effet électro-attracteur)
- Une densité électronique moins grande
- Un champ magnétique ressenti plus élevé (proton déblindé)
- Une fréquence de résonance plus élevée
- Un signal déplacé vers la gauche

7.7 L’intégration des pics

L’aire sous le pic est proportionnelle au nombre de protons représentés par chaque pic.
7.8 Les régions du spectre de RMN du Proton
Déplacements de référence: CH3 (0.9 ppm), CH2 (1.3 ppm), CH (1.7 ppm)
Les groupes électro-attracteurs déplacent ces signaux de 1 à 3 ppm
Remarque: Les effets sont additifs
7.9 Le couplage: interaction entre H voisins
Permet de comprendre la structure du squelette hydrocarboné
Considérons 2 protons d’une même molécule: HA et HX
7.10 La constante de couplage en Hz
7.11 Le couplage avec 2 H voisins
7.12 Construire simplement les signaux
7.13 Le triangle de Pascal
7.14 Exemples de spectres
7.15 Constantes de couplage typiques
Les constantes de couplage dépendent de 3 facteurs
- La distance entre les protons à travers les liaisons: 2J, 3J, 4J - liaison simple ou double
- L’angle entre les deux liaisons C-H: libre rotation, cis, trans
- Les substituants électronégatifs: diminution de la constante de couplage