Catalyse redox
3 Les cytochromes de la chaine de transport des électrons
L’oxygène est un bon agent oxydant mais potentiellement dangereux. Si la cellule doit l’utiliser dans un métabolisme, une distance raisonnable doit exister entre les sites que l’O2 doit oxyder et les diverses réactions métaboliques. Cet isolement est réalisé par une série de protéines de potentiel standard décroissant, qui font passer les électrons à travers l’oxygène dans les mitochondries de la cellule. Une version schématique du transport d’électron des mitochondries est proposée sur la figure suivante.
ADP ADP ADP
NAD+ FADH2 Quinone 2Fe(II) 2Fe(III) 2Fe(II) 2Fe(III) H2O
Coenxyme Cyt b Cyt c1 Cyt c Cyt aa3
NADH FAD Hydroquinone 2Fe(III) 2Fe(II) 2Fe(III) 2Fe(II) 1/2O2
ATP ATP ATP
-0,28 -0,22 -O,08 -0,05 +0,22 +0,25 +0,28 +0,81
Eø (pH=7)/V
Séquence des réactions de la chaîne de transport électronique dans les mitochondries
Les participants les plus remarquables sont les cytochromes, un groupe d’hème-protéines ayant Fe dans un ligand de type porphyrine.
On trouve des enthalpies libres de réaction élevées à deux endroits de la figure 1, l’une pour la réaction entre la paire de cytochromes notés cyt b et cyt c1, l’autre là où le cytochrome a réagit avec l’oxygène (cytaa3 et O2). L’énergie libérée dans ces étapes peut être stockée pour une utilisation ultérieure dans la cellule en couplant la réaction avec la formation de ATP à partir de l’ADP et de HPO42-. Cette chaine peut paraître inutilement complexe, mais elle fragmente la réaction de O2 avec NADH en des étapes discrètes prudentes. Cette séquence permet un couplage avec la synthèse de plusieurs molécules d’ATP ainsi que l’organisation spatiale du processus d’un côté de la membrane (où est localisé la cytochrome) à l’autre.
Les cytochromes dans la chaine de transport des électrons sont des protéines qui réduisent O2 étape par étape.
a) Caractère de sphère externe
Les cytochromes fonctionnent en faisant faire au fer la navette entre les Fe(II) et les Fe(III) sur le site actif, sont des agents de transfert monoélectronique. Les atomes de fer sont coordinés à des cycles porphyrine enfouis au milieu de la protéine. Grâce à la modélisation informatique, on peut générer des modèles plausibles du complexe qui se forme lorsque deux de ces protéines se rencontrent pour échanger un électron. Comme les atomes Fe des deux complexes qui se rencontrent restent toujours très éloignés l’un de l’autre, la réaction ne peut pas être un cas de transfert électronique de sphère interne. Elle doit se produire par un transfert électronique à longue distance de sphère externe.
Le tableau 2. donne des constantes de vitesse, des potentiels standard et les distances minimales d’approche entre le bord de la porphyrine contenant l’atome de fer et son partenaire dans le transfert électronique. Il est instructif de comparer ces paramètres protéine-protéine avec des paramètres similaires pour les réactions d’une protéine avec des complexes métalliques simples, et on a également inclus dans le tableau certains d’entre eux. La variation de la vitesse de réaction avec la distance et l’énergie est particulièrement intéressante.
Tableau 2 : Quelques vitesses des réactions de transfert électronique de protéines rédox.
|
Couple |
E/V |
R/Å |
k/s-1 |
|
FeIIcytb5/FeIIIcytc |
0,2 |
8 |
1,5.103 |
|
Znapocytc*/FeIIb5 |
0,8 |
8 |
3.105 |
|
H2porfc*/FeIIIb5 |
0,4 |
8 |
1.104 |
|
Zncytb5/FeIIIcytc |
1,1 |
8 |
1.103 |
|
RuHis33/cytc |
0,15 |
11 |
40 |
|
RuHis/azurin |
0,2 |
10 |
2,5 |
|
RuHis/Mb |
0,05 |
13 |
0,02 |
|
FeIIccp/FeIIIcytc |
0,4 |
16 |
0,025 |
|
FeIIIccp/porfcytc |
1,0 |
16 |
180 |
La plupart des abréviations désignent des protéines. Elles sont élaborées dans les textes biochimiques. Nous n’avons pas besoin ici d’en connaître les détails. Tout ce que signifient ici les différentes abréviations, c’est que les différents membres de la famille des protéines de transfert électronique sont associés à des valeurs de Eø, et RuHis se rapporte au groupement (NH3)5Ru fixé à une histidine (dont le numéro est indiqué si nécessaire) de l’enzyme. On peut faire varier les distances en changeant l’unité histidine à laquelle le Ru est fixé. La notation* indique une réaction utilisant un état photoexcité pour rendre la réaction plus exogonique.
L’oxydation et la réduction du fer des cytochromes semblent être contrôlées par des transferts électroniques de sphère externe.
b) Effet de la distance et effet tunnel
L’influence de la distance sur la constante de vitesse est souvent interprétée par l’effet tunnel quantique, selon laquelle une particule peut traverser une barrière bien que son énergie soit insuffisante pour la surmonter. Dans les modèles simples de l’effet Tunnel, la probabilité de trouver la particule au-delà de la barrière décroit exponentiellement lorsque la largueur de la barrière augmente. Les données du tableau 2 confirment que la distance entre les centres métalliques a l’effet prévu si on limite la comparaison aux structures similaires.
Les vitesses de transfert électronique entre les sites rédox décroissent à peu près exponentiellement lorsque la distance augmente.
c) Influence de l’énergie et théorie de Marcus
On peut examiner la variation de la vitesse de réaction avec l’énergie à la lumière de la théorie de Marcus.
ĸ2 = fk1k2K
k1 et k2 sont les constantes de vitesse de réaction des deux réactions d’échange ;
K est la constante d’équilibre de l’ensemble de la réaction ;
f est un paramètre composite des constantes de vitesse et de la vitesse de collision : on peut le prendre proche de 1 pour les calculs approximatifs.
Avec f ~ 1, 2lnkt = lnk1 + lnk2 + lnK
L’équation de Marcus peut ainsi s’exprimer comme relation linéaire des énergies libres parce que les logarithmes des constantes sont proportionnels aux énergies de Gibbs d’activation.
2∆G = ∆G1 + ∆G2 + ∆tGø
lnkt2 = lnk1k2 - ∆tGø/2RT
Le produit k1k2 des constantes de vitesse d’auto-échange reflète la barrière intrinsèque du transfert électronique entre les deux complexes. La constante d’équilibre K est une mesure de l’enthalpie libre de la réaction globale. Malheureusement, il est difficile d’estimer la barrière intrinsèque pour de nombreuses protéines, parce qu’il n’est pas possible d’étudier l’auto-échange entre deux degrés d’oxydation d’une protéine.
La variation de kT avec l’enthalpie libre de la réaction globale (et, de façon équivalente, avec les potentiels standards des protéines, car ∆TGø = -vFEø) est également ce que prévoit la théorie de Marcus pour des protéines de structures similaires. Toutefois, la structure joue un rôle dans la détermination de kT, et les constantes de vitesse des protéines ayant des structures différentes peuvent différer de plusieurs ordres de grandeur. Par exemples, les trois premiers composés du tableau I.16 forment une série où la dépendance par rapport à Eø est celle prédite par la théorie de Marcus. De même, les deux derniers composés peuvent représenter un couple de structures similaires. Le rôle de la structure de l’enzyme dans la détermination de la barrière intrinsèque apparaît clairement lorsqu’on compare le quatrième composé aux trois premiers.
La conclusion générale semble être que les cytochromes et plusieurs autres protéines rédox sont de simples réactifs de transfert monoélectronique de sphère externe.
Pour les sites redox avec des structures similaires, les constantes redox diminuent avec la décroissance de l’énergie standard de réaction de Gibbs.
d) Le cytochrome P-450
Le cytochrome P-450 est le nom générique d’une famille d’enzymes ayant comme sites actifs des porphyrines de fer qui catalysent l’addition de l’oxygène à un substrat hydrocarboné. La désignation « P-450 » vient de la position de la bande d’absorption caractéristique du bleu à l’ultraviolet proche, de la porphyrine ; cette bande est appelée « bande de Soret » ; elle est déplacée vers le rouge jusqu’à 450nm pour les complexes carbonyles de ces molécules. La réaction représentative la plus importante de cette catégorie de réactions est la réaction d’insertion :
RH + 1/2O2 ------> ROH
L’insertion de O dans une liaison RH qui est exactement le type de réaction rédox qui devrait se produire par transfert d’atome – fait partie de la défense du corps contre les composés hydrophobes comme les médicaments, les précurseurs stéroïdes et les pesticides. L’hydroxylation de RH en ROH rend ces composés plus solubles dans l’eau et facilite ainsi leur élimination.
Le cycle catalytique proposé pour P-450 est le cycle 2 la séquence commence en (a), l’enzyme étant au repos avec le fer sous forme de Fe(III). Le substrat hydrocarboné se fixe ensuite (b), puis un transfert monoélectronique vers la porphyrine de fer donne (c). Ce complexe de Fe(II) avec le substrat lié fixe O2 en produisant (e). (Á ce point du cycle, une réaction compétitive avec CO qui donne (d) conduit à une espèce aisément identifiable qui est responsable de l’absorption à 450 nm qui donne son nom à l’enzyme). Une réaction clé est la réduction du cycle porphyrinique du complexe oxygéné (e) par un second électron, qui donne l’anion radical du cycle. La capture des deux ions H+ conduit ensuite à la formation du complexe oxo de Fe(IV) (f) qui attaque le substrat hydrocarboné pour insérer l’oxygène. La perte de ROH et la capture d’une molécule H2O sur la position de coordination vacante ramène le cycle à l’état de repos. La clé du cycle est la formation du complexe oxo de Fe (IV) (f).
Le cycle catalytique proposé pour p-450 est le cycle 2.
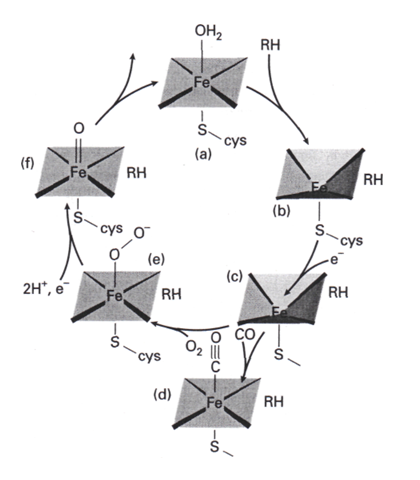
(a) Structure du site actif
La séquence d’aminoacide de la protéine crée une structure plissée autour du sité hème et de son atome Fe, formant un environnement protégé de la solution et de faible permittivité. Le substrat hydrocarboné est fixé tout près, et la liaison C—H attaquée doit être à moins de 5 Å du site de Fe. Le ligand de l’atome Fe situé en trans par rapport au site de fixation de O2 est la chaîne latérale thiolate d’un résidu cystéine (14). Lorsque O2 se coordine, l’extrémité extérieure de la molécule O2 se projette dans la solution, mais le substrat est fixé à l’enzyme à l’intérieur de la poche hydrophobe.
L’atome de fer du site actif de P-450 est protégé par des substituants hydrocarbonés, mais O2 se projette dans la phase aqueuse
(b) Mécanisme d’oxygénation
Le mécanisme précis de l’oxygénation par P-450 reste le sujet de recherches intensives. On pense qu’il y a deux possibilités, l’une impliquant la formation d’une espèce radicalaire d’oxygène qui peut attaquer la liaison C—H, et l’autre le transfert d’un atome O vers la liaison C—H. Il n’y a pas de bons précédents inorganiques pour l’oxydation d’un composé organique par Fe(IV)O, et le débat actuel sur le mécanisme de la réaction P-450 élargira presque certainement pour les chimistes inorganiciens la gamme des mécanismes d’oxydation possibles. La proposition d’un mécanisme radicalaires sont peu sélectives et ne sont pas stéréospécifiques ; les oxydations de P-450 sont très sélectives et conservent l’activité optique des substrats chiraux.
La proposition selon laquelle l’oxygénation se produit par l’attaque d’un centre oxygène positif sur la liaison C—H n’est qu’un exemple d’un nouveau mécanisme proposé pour expliquer une réaction enzymatique. Il est corroboré par l’observation de la rétention de la configuration stéréochimique et par la diminution de la réactivité lorsque le substrat possède des substituants qui empêchent l’atome O de s’approcher de la liaison C—H. Le transfert de ce qui est en fait un atome O neutre laisse la porphyrine de fer réduite de Fe(IV) à Fe(III), alors que le substrat organique est oxydé.
Le rôle de l’atome S dans cette séquence peut s’exprimer en termes d’orbitales moléculaires en concentrant notre attention sur les orbitales atomiques du fragment S—Fe---O. Si nous limitons notre attention sur les orbitales 3pz du soufre, 3dz2, 3dzx, 3dyz du fer et 2p de l’oxygène, nous obtenons le diagramme d’orbitales. Les 13 électrons disponibles du fragment occupent les niveaux liants, les niveaux faiblement antiliants, et le dernier électron occupe l’orbitale fortement antiliante 3σ. On peut donc rationaliser la charge formelle élevée du fer dans le complexe par la stabilisation due à l’importante donation de charge de l’atome S très polarisable vers les orbitales 2σ et 2π du fragment. Le rôle particulier de l’atome S trans est de contrôler la charge de l’atome Fe de façon à ce qu’un atome O puisse être transféré.
Il apparaît que l’atome de soufre coordiné de P-450 contrôle la charge du fer, facilitant ainsi le transfert de l’atome O.